
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿 女,4岁,因"发热8天,鼻塞流涕5天"入院。8天前患儿无明显诱因出现发热,热峰39.3℃,口服退热药可降至正常,间隔6~8 h易复升。2018年1月21日至2018年2月26日因"肺脓肿、肺炎"在我院胸心外科行手术治疗,已愈。1年前曾先后因"下肢脓肿、化脓性腮腺炎"行引流术后治愈。1月前因"血性痰液"住院治疗,期间无发热,血性痰渐消失,已排除"肺结核及肺脓肿",已愈。查体:患儿右耳道可见脓性分泌物,鼓膜窥及处充血、浑浊。左、右侧颈部可触及肿大的淋巴结,无触痛。既往反复头部湿疹史,有鼻窦炎病史。临床怀疑免疫缺陷。父母体健,非近亲结婚,有1个妹妹,3岁,体健。
实验室检查:血常规示白细胞15.48×109/L(偏高),红细胞3.95×1012/L,血红蛋白113 g/L,血小板230×109/L。中性粒细胞百分比62.7%,淋巴细胞百分比29.3%,单核细胞百分比4.6%,中性粒细胞计数9.71×109/L(偏高),淋巴细胞计数4.53×109/L(偏高),单核细胞计数0.71×109/L, CRP 93.69 mg/L(偏高)。白细胞增高,以中性粒计数增高为主,CRP增高,提示存在细菌感染。免疫球蛋白E定量>6000.000 ng/mL,明显升高;降钙素原0.027 ng/mL(正常);白介素IL-6 92.27 pg/mL(偏高);淋巴细胞分型计数:T淋巴细胞73.36%, T8淋巴细胞25.83 %, T4淋巴细胞46.45 %, CD4/CD8 1.80, NK淋巴细胞1.79%(偏低),B淋巴细胞23.82%,总淋巴细胞绝对计数3849.29/μL(偏高),T淋巴细胞绝对计数2823.99/μL, T8淋巴细胞绝对计数994.15/μL, T4淋巴细胞绝对计数1787.85/μL(偏高), NK淋巴细胞绝对计数69.08/μL(偏低),B淋巴细胞绝对计数916.95/μL(偏高),双阴性T淋巴细胞百分数1.08%,双阳性T淋巴细胞百分数0.84 %,双阴性T淋巴细胞数41.99 U/L,双阳性T淋巴细胞数32.51 U/L(偏高),提示存在细胞免疫功能紊乱。粒细胞呼吸爆发试验:血细胞簇分化抗原(CD45)检测阳性(+),粒细胞活化率-PBS刺激2.6 %,粒细胞活化率-PMA刺激95.4 %,无异常。肝肾功能、电解质正常。
辅助检查:心电图示异位心律,房性自主心律,非特异性室内传导异常,部分导联T波异常。胸部CT示双肺纹理粗,肺野透光度不均,左上叶尖后段可见类圆形囊状低密度影,壁略厚,后缘邻近叶间胸膜增厚;余两肺上下叶可见斑片及条片影。颈部淋巴结彩超:双侧颈部可见数个边界清晰、大小不等的淋巴结回声堆积。耳纤维内镜:左耳道通畅,鼓膜充血,右耳外耳道肿胀充血可见大量分泌物,鼓膜不能窥及。16排CT头颅+副鼻窦平扫示:鼻窦炎,双侧下鼻甲肥大,双侧鼻腔及双侧后鼻孔区、右侧鼻前庭可见软组织密度影充填,腺样体肥大。
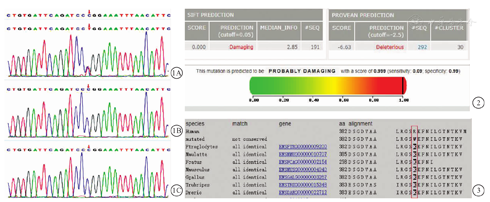
全外显子高通量测序并Sanger测序验证:为进一步确诊,经患儿父母签署知情同意书,并通过本院医学伦理委员会批准(YZ-19-117号)后,抽取患儿及父母静脉血样,提取样本DNA。用美国IDT公司xGen® Exome Research Panel v1.0捕获探针与gDNA文库序列进行液体杂交,将目标区域DNA片段进行富集,构建全外显子文库。通过Illumina公司NovaSeq 6000系列测序仪进行高通量测序(PE150),筛选出的可疑变异位点,设计引物,并对患儿及其父母变异位点采用Sanger测序进行验证,并确定遗传来源。基因检测结果如图1所示,患儿信号转导与转录活化因子3(signal transducers and activators of transcription 3, STAT3)基因有1个杂合变异(位于17号染色体):转录本号NM_139276,第13外显子:c.1144C>T,导致氨基酸改变p.R382W (p.Arg382Trp),为错义突变;患儿的父亲、母亲该位点均无变异,此变异为新发突变。在100名正常对照者中未检测到该突变。应用蛋白功能预测软件SIFT、PROVEAN、PolyPhen2预测突变位点,预测值分别为0.0、-6.63、0.99,均为"有害""有害""可能有害"(图2)。应用Mutation Taster物种保守性分析显示STAT3基因p.R382W位点氨基酸在不同物种间几乎完全相同,表明在进化上高度保守(图3)。根据美国医学遗传学和基因组学学会指南,该变异判定为致病性变异。
高IgE综合征(hyper-IgE syndrome, HIES),又称Job综合征,是一种临床罕见的原发性免疫缺陷病,多以常染色体显性遗传为主。该病以反复的肺、皮肤金黄色葡萄球菌感染为突出特点[2]。在婴幼儿阶段常出现葡萄球菌复发性皮肤感染及皮肤黏膜念珠菌感染和细菌性肺炎,肺囊肿和支气管扩张通常是由于肺炎的迁延所致。在新生儿期常出现皮疹,随后发展为湿疹样皮炎。非免疫性特征包括保留的乳牙,脊柱侧弯,轻微创伤后的骨折,关节过度伸展和特征性的面部外观[3],鼻宽度增加,上颚高,通常在青春期出现。成人期的症状包括血管异常(动脉瘤、心肌梗死、蛛网膜下腔出血),胃肠道异常(包括胃食管反流病、食道运动障碍和自发性肠穿孔及胃肠道的真菌感染[4]),淋巴瘤的发生率增加。实验室检查结果显示:血清免疫球蛋白E(IgE)浓度升高至2000IU /mL以上(成人正常<100 IU/ mL),嗜酸性粒细胞增多(>700/μL),T和B细胞减少,几乎没有产生IL-17的Th17细胞[5]。患者治疗不当或不及时引起严重的肺部感染及其他并发症是致死的主要原因。该病临床表现复杂,容易误诊和漏诊,基因检测有助于早期诊断和治疗。
2007年首次明确STAT3基因为HIES的致病基因[5],STAT3基因位于染色体17q21区,共包含23个外显子,编码770个氨基酸的STAT3蛋白,包括N端保守序列、卷曲螺旋区、DNA结合域(DBD)、连接区、SH2区和转录活性区域,是Th17细胞发育必需的转录因子及多种细胞因子( IL-6、IL-10、IL-22、IL-23等)信号途径的中心因子[6]。急性炎症因子IL-6信号通路受损或许可以解释HIES"冷脓肿"。Th17细胞辅助T细胞,对于清除真菌和细胞外细菌感染至关重要[7]。Th17分化发育障碍使患者对金黄色葡萄球菌和真菌的免疫功能受损[8],无法产生Th17细胞是潜在的HIES对反复感染易感性的基础,不能分泌IL-17来抵御胞外菌和真菌,金黄色葡萄球菌肺炎易并发肺脓肿、肺大泡和气胸,危及生命。
STAT3基因突变的显性负效应是导致常染色体显性遗传的高IgE综合征的主要原因[9]。目前HGMD数据库报道的STAT3基因变异有100余种,包括错义突变、无义突变、剪接突变、大片段的缺失等。突变的STAT3与野生STAT3共表达发挥着显著负性的作用,部分抑制正常蛋白功能。大多数HIES患者至少一条STAT3等位基因存在单一位点突变,其产生的突变蛋白会对另一个正常等位基因表达的STAT3蛋白产生显性负效应,导致STAT3蛋白功能紊乱,最终导致HIES发生。虽然多数STAT3基因缺乏或突变表现为HIES,但表现仍呈临床异质性,携带相同突变的同一家系成员,其临床感染程度、免疫性疾病和体征等也各异[10]。STAT3基因突变患者也存在NK细胞功能和数量的缺陷[11],并伴随Thl7细胞的缺乏和IL-17的产生障碍,导致角质形成细胞及气道上皮细胞中性粒细胞趋化功能减弱,分泌的抗菌肽减少,故HIES患者感染好发于皮肤及呼吸道。口腔黏膜分泌的IL-17能诱导产生唾液中的富组蛋白,富组蛋白具有抗真菌活性,其缺乏导致HIES患者易患口腔念珠菌病。
本例患儿有反复肺脓肿、肺炎、头部湿疹史,既往腮腺炎、鼻窦炎病史;免疫球蛋白E定量明显升高,白介素IL-6升高,淋巴细胞分型计数提示存在细胞免疫功能紊乱。临床高度怀疑为免疫缺陷性疾病,进一步行全外显子测序,结果显示其STAT3基因存在c.1144C>T杂合突变,导致第382号氨基酸由精氨酸变异为色氨酸,gnomAD东亚人群频率为未知。我们在100名正常对照者中未检测到上述位点突变,可排除单核苷酸多态性的可能性。经蛋白功能预测软件预测,上述变异可能导致蛋白质功能受影响。经Sanger测序验证,该变异父母均为野生型,为自发突变。经查阅文献,结合临床表现和辅助检查,提示该患儿可能是STAT3基因变异导致的高IgE综合征。目前该病尚无针对性治疗方法,患儿先后予头孢呋辛及头孢他啶针联合阿奇霉素抗感染,补液及雾化等综合治疗,呼吸道及其他系统感染症状明显改善。
综上所述,对于长期反复不明原因的感染,疑似免疫缺陷患儿,应积极进行病因调查,基因检测有助于早期诊断。本病例的诊治对进一步探讨STAT3基因变异机制,研究基因型-表型相关性和指导疾病的诊疗预后有着重要意义,拓展了STAT3基因的变异谱,为高IgE综合征遗传分析及临床诊断积累了经验。
所有作者均声明不存在利益冲突

























