
巨脑畸形-多小脑回-多指(趾)畸形-脑积水综合征是一种罕见的常染色体显性遗传病,无特效治疗方法。本文报道1例出生即发现特殊外貌、头围增大、六指畸形的患儿,为国内首次报道CCND2基因突变所致病例,为早期脑积水的诊断拓宽思路。


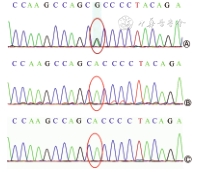
患儿男,生后5 h,因“发现低血糖3 h”入院。患儿胎龄39+6周顺产娩出,出生体重2 900 g,出生史无异常,生后2 h常规监测发现血糖低。母孕28周时胎儿超声提示韦式腔及双手六指畸形。入院查体:特殊外貌,头围38 cm(>3SD),前额突出、眼距宽、耳位低、乳距宽,双手六指畸形(图1),双侧隐睾,心肺腹无异常。辅助检查:TORCH核酸及IgG抗体阴性。血糖1.6 mmol/L。头颅彩超示双侧脑室增宽,右侧13.5 mm,左侧11.8 mm。脑脊液生化:蛋白1.66 g/L,氯128.7 mmol/L。血培养、脑脊液培养阴性。眼底未见异常,双侧听力筛查未通过。头颅磁共振成像示巨脑畸形、多小脑回,伴梗阻性脑积水。入院后予对症支持治疗,血糖正常,能自行纳奶,生后第12天出院。患儿外显子高通量测序结果显示:巨脑畸形-多小脑回-多指(趾)畸形-脑积水(megalencephaly-polymicrogyria-polydactyly-hydrocephalus,MPPH)综合征相关基因CCND2发现1处杂合突变c.838A>G,导致氨基酸改变p.Thr280Ala,父母此位点基因型均正常,见图2。生后6个月患儿出现抽搐,脑电图提示右侧显著高度失律,右侧半球多灶性棘波、(多)棘慢波发放,监测到1次成串痉挛发作,诊断癫痫,予左乙拉西坦治疗后未再出现明显抽搐发作。1.5岁时随访,患儿头围47 cm(P25~P50),体重9 kg(P3),身高78 cm(P3~P10),仅能说单音节字,不能进行简单语言交流,仅会抬头,不会翻身、爬、坐。
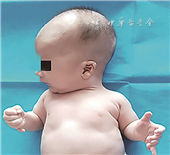
MPPH综合征是一种罕见的常染色体显性遗传病,其特征是脑膨大、大脑皮层畸形、多小脑回,该综合征首次报道于2004年[1]。MPPH综合征患儿脑室均有不同程度的增大,近50%的患儿合并脑积水;均存在运动、智力障碍,约50%的患儿合并癫痫、轴后六指(即第6指出现在尺侧)。2012年美国医学遗传学杂志发表的MPPH综合征诊断标准为,当患者有核心症状中的①+②,但不伴有血管异常、并指或异位可诊断MPPH:(1)核心症状:①早期过度生长(脑>躯体组织),早发的巨脑畸形(头围>2SD);②大脑结构畸形/多小脑回;③远端肢体异常,多指。(2)支持症状:选择性脑过度生长;脑室扩大/脑积水;小脑扁桃体异位;胼胝体异常;肌张力低下,发育落后,独特的面部特征如额头凸、长头畸形等[2]。患儿还可有喂养困难、视觉问题、先天性心血管畸形、甲状腺疾病、重复肾以及明显的低血糖等[3, 4]。本例患儿出生时有特殊外貌,头围增大(>3SD),合并多指、脑室增宽、巨颅畸形、多小脑回、脑积水,临床诊断MPPH综合征明确。
MPPH综合征由AKT3、CCND2、PIK3R2基因的致病性突变所致。本例患儿全外显子高通量测序发现CCND2基因错义突变,综合患儿临床资料、影像学资料及基因检测结果诊断为MPPH综合征,后期随访出现癫痫、严重精神、运动发育落后、智力低下,也符合MPPH综合征。目前MPPH综合征无特效治疗,以对症治疗为主。
所有作者均声明不存在利益冲突

























