
探讨儿童颅内非典型畸胎样/横纹肌样瘤(AT/RT)的诊断、治疗策略及预后。
回顾性分析2012年1月至2019年6月郑州大学第一附属医院经病理学诊断为颅内AT/RT的15例患儿的临床资料,采用Kaplan-Meier法计算总体生存(OS)率和无进展生存(PFS)率。采用Log-rank比较不同组间生存差异,采用COX回归模型研究影响生存的因素。
本组患儿男12例,女3例;中位年龄5.5岁(8个月~17.1岁);患儿均首选手术切除,其中全切10例,次全切5例;6例患儿行"手术+放疗+化疗+鞘内注射",4例患儿行"手术+化疗+鞘内注射",2例患儿行"手术+放疗",3例患儿仅行手术治疗。截至2020年1月,15例患儿中位生存期为18个月(1~27个月),存活率为33.3%。患儿1年OS率和PFS率分别为71.5%和49.7%,2年OS率和PFS率分别为17.9%和0。Log-rank检验结果显示<3岁和≥3岁患儿1年OS率分别为87.5%和57.1%,差异有统计学意义(χ2=4.257,P=0.039)。全切组和次全切组患儿1年OS率分别为90.0%和40.0%,差异有统计学意义(χ2=6.057,P=0.014)。肿瘤未播散组和播散组患儿1年OS率分别为100.0%和33.3%,差异有统计学意义(χ2=9.865,P=0.002)。标危组和高危组患儿1年OS率分别为88.9%和41.7%,差异有统计学意义(χ2=5.111,P=0.024)。COX回归模型显示,年龄、肿瘤切除程度、肿瘤是否播散和危险度分层是影响患儿OS预后的独立危险因素[风险比(HR)=3.411、3.795、5.245、3.397,P=0.025、0.011、0.001、0.017]。
儿童颅内AT/RT临床罕见,初诊困难,预后差,最大范围的安全切除仍是首选治疗,年龄、肿瘤切除程度、肿瘤是否播散和危险度分层是AT/RT患儿的独立预后因素。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
颅内非典型畸胎样/横纹肌样瘤(atypical teratoid/rhabdoid tumor,AT/RT)是一种罕见的高度恶性的中枢神经系统胚胎性肿瘤,占所有儿童颅内肿瘤的1%~2%[1]。根据2016年世界卫生组织(WHO)中枢神经系统肿瘤分类,为WHO Ⅳ级的胚胎性肿瘤[2]。超过90%的AT/RT发生于3岁以下的婴幼儿,在<3岁的儿童脑肿瘤中约占20%[3]。20%~40%的患儿在第1次诊断时就发生了肿瘤播散/转移,中位生存期12~17个月,预后极差[4]。由于颅内AT/RT罕见,目前尚无统一标准的治疗方案。本研究对郑州大学第一附属医院神经外科2012年1月至2019年6月收治的15例颅内AT/RT患儿的诊治及预后进行回顾性研究,以期对此类疾病的诊治提供参考及帮助。
2012年1月至2019年6月郑州大学第一附属医院神经外科共收治初次诊断颅内AT/RT患儿15例,均经磁共振成像(MRI)和病理学明确诊断。患儿监护人均自愿接受治疗,并签署知情同意书,本研究通过郑州大学第一附属医院医学伦理委员会批准(批准文号:2020-KY-155)。
本中心根据于患儿年龄及危险分层对儿童AT/RT采取个体化治疗。肿瘤切除程度可分为全切除(术后MRI提示肿瘤无残余),次全切除(术后MRI提示肿瘤切除>90%)。根据肿瘤切除程度、影像学和脑脊液检查结果将患儿分为标危组和高危组。满足以下条件者为标危组:肿瘤完全切除,影像学提示无中枢神经系统或颅外播散,腰椎穿刺脑脊液未发现肿瘤细胞。满足以下条件者为高危组:肿瘤部分切除,影像学及脑脊液细胞学检查提示中枢神经系统或颅外播散。
患儿均行显微外科手术切除,手术切除策略:(1)肿瘤全切除(肿瘤位于非功能区);(2)最大安全范围切除肿瘤(肿瘤累及功能区)。
(1)初诊年龄>3岁的患儿:术后4周开始放疗,对于标危患儿,行局部瘤床三维适形放疗,照射剂量54~55 Gy;对于高危患儿,行局部瘤床54~55 Gy放疗,全脑和全脊髓36 Gy放疗。(2)初诊年龄≤3岁的患儿:标危患儿不行放疗;高危患儿延迟至3岁后放疗或化疗后行局部瘤床放疗。如果化疗结束年龄未达3岁,可行局部瘤床放疗,3岁后也不行全中枢放疗。
对于初诊年龄≥3岁的患儿,放射治疗结束后4周开始化疗;对于初诊年龄<3岁的患儿,因不行放射治疗,则在术后2~4周开始化疗,化疗方案为"CTX+DDP+VCR":环磷酰胺(CTX,750 mg/m2,第1-2天)+顺铂(DDP,75 mg/m2,第1天)+长春新碱(VCR,1.5 mg/m2,第1、8天),同步水化3 000 mL/(m2·24 h),每4周重复,共8个疗程。
在全身化疗的基础上,每次化疗前行腰椎穿刺鞘内注射:甲氨蝶呤(MTX,10 mg/m2)+阿糖胞苷(Ara-c,30 mg/m2)+地塞米松(Dex,2.5~5.0 mg),共7次。对于影像学及脑脊液细胞学检查提示中枢神经系统肿瘤播散的患儿,可继续予鞘内注射药物应用,最多不超过20次。
通过门诊、电话对患儿进行随访。治疗期间每次化疗前行全脑脊髓MRI和脑脊液细胞学检查判断有无播散转移,治疗结束后每3~6个月复查MRI。终点事件定义为复发与否,通过临床和影像学检查评判复发,随访时影像学检查发现肿瘤全切后患儿原手术部位再次出现肿瘤生长或大部分切除患儿残余病灶组织较之前增大定义为复发。无进展生存期为手术次日至复发的时间,失访或随访结束未有复发的作为结尾值。
采用SPSS 22.0软件对数据进行分析。符合正态分布的计量资料以 ±s表示,非正态计量资料采用中位数(P25,P75)表示。存活率为随访截止点存活例数占总体随访患儿的百分比,采用Kaplan-Meier法计算患儿的总生存(OS)率和无进展生存(PFS)率,2组间生存比较采用Log-rank检验,采用COX回归模型研究影响生存的多种因素,P<0.05为差异有统计学意义。
±s表示,非正态计量资料采用中位数(P25,P75)表示。存活率为随访截止点存活例数占总体随访患儿的百分比,采用Kaplan-Meier法计算患儿的总生存(OS)率和无进展生存(PFS)率,2组间生存比较采用Log-rank检验,采用COX回归模型研究影响生存的多种因素,P<0.05为差异有统计学意义。
本组患儿共15例。男12例,女3例,男女比例为4:1;年龄8个月~17.1岁,中位年龄5.5岁,3岁以下的患儿7例(46.7%)。主要临床表现:首诊症状为颅内压增高者12例,局灶神经功能缺损症状者2例,癫痫1例(表1)。

颅内非典型畸胎样/横纹肌样瘤15例患儿临床资料
Clinical data of 15 cases with intracranial atypical teratoid/rhabdoid tumor
颅内非典型畸胎样/横纹肌样瘤15例患儿临床资料
Clinical data of 15 cases with intracranial atypical teratoid/rhabdoid tumor
| 临床特点 | 例数(%) | |
|---|---|---|
| 年龄 | ||
| <3岁 | 7(46.7) | |
| ≥3岁 | 8(53.3) | |
| 性别 | ||
| 男 | 12(80.0) | |
| 女 | 3(20.0) | |
| 首发症状 | ||
| 颅高压 | 12(80.0) | |
| 神经功能缺损 | 2(13.3) | |
| 癫痫 | 1(6.7) | |
| 肿瘤位置 | ||
| 幕上 | 8(53.3) | |
| 幕下 | 7(46.7) | |
| 肿瘤大小 | ||
| <5 cm | 6(40.0) | |
| ≥5 cm | 9(60.0) | |
| 梗阻性脑积水 | ||
| 有 | 6(40.0) | |
| 无 | 9(60.0) | |
| 切除程度 | ||
| 全切除 | 10(66.7) | |
| 次全切除 | 5(33.3) | |
| 肿瘤播散(转移) | ||
| 无播散 | 9(60.0) | |
| 中枢神经系统播散 | 5(33.3) | |
| 转移(肺) | 1(6.7) | |
| 病理表现 | ||
| 波形蛋白(+) | 15(100.0) | |
| 上皮膜抗原(+) | 13(86.7) | |
| S-100(+) | 11(73.3) | |
| 角蛋白(+) | 10(66.7) | |
| 胶质纤维酸性蛋白(+) | 8(53.3) | |
| INI1(+) | 0(0) | |
| 治疗 | ||
| 手术+放疗+化疗+鞘内注射 | 6(40.0) | |
| 手术+化疗+鞘内注射 | 4(26.7) | |
| 手术+放疗 | 2(13.3) | |
| 手术 | 3(20.0) | |
15例患儿术前均行CT及MRI检查。根据肿瘤位置分类,肿瘤位于幕上者8例,位于幕下者7例。肿瘤体积<5 cm者6例,≥5 cm者9例。术前合并梗阻性脑积水6例。CT扫描可见病灶呈等密度10例,呈高密度5例,其中6例可见病灶内低密度坏死或囊变区域,2例病灶内见高密度钙化,1例病灶合并出血。增强MRI呈多样性,肿瘤实质部分不均匀强化12例,明显均匀强化2例,轻度强化1例。
本组15例肿瘤均有INI1蛋白表达的缺失,结蛋白(Desmin)和肌浆蛋白(Myogenin)表达均为阴性,波形蛋白(Vimentin)、胶质纤维酸性蛋白(GFAP)、上皮膜抗原(EMA)、突触体素(SYN)、角蛋白(CK)和S-100不同程度表达阳性,Ki-67阳性指数为30%~70%(表1)。
15例患儿均行显微外科手术切除,肿瘤全切除10例(66.7%),肿瘤次全切除5例(33.3%)。脑脊髓MRI和脑脊液细胞学检查提示肿瘤无播散9例,中枢神经系统播散5例,1例患儿经胸部CT及全身正电子发射计算机断层扫描(PET-CT)检查确诊肺部多发转移。对患儿进行危险分层,其中标危组9例,高危组6例。结合患儿一般情况、年龄及家属治疗意愿,治疗方式见表1。
截止2020年1月,5例患儿存活(存活率为33.3%),其中,3例患儿肿瘤无进展或复发,2例患儿肿瘤原位复发;10例已死亡的患儿中,8例因肿瘤复发死亡,2例因术后并发症死亡(1例术后颅内出血,1例术后颅内感染)。中位随访时间10个月,15例患儿中位生存期为18个月(1~27个月)。患儿1年OS率和PFS率分别为71.5%和49.7%,2年OS率和PFS率分别为17.9%和0(图1)。

注:AT/RT:颅内非典型畸胎样/横纹肌样瘤;OS:总生存;PFS:无进展生存 AT/RT:intracranial atypical teratoid/rhabdoid tumor;OS:overall survival;PFS:progression free survival
Log-rank检验结果显示,<3岁和≥3岁患儿1年OS率分别为87.5%和57.1%,差异有统计学意义(χ2=4.257,P=0.039)。全切组和次全切组患儿1年OS率分别为90.0%和40.0%,差异有统计学意义(χ2=6.057,P=0.014)。肿瘤未播散组和播散组患儿1年OS率分别为100.0%和33.3%,差异有统计学意义(χ2=9.865,P=0.002)。标危组和高危组患儿1年OS率分别为88.9%和41.7%,差异有统计学意义(χ2=5.111,P=0.024)。COX回归分析结果显示,年龄、肿瘤切除程度、肿瘤是否播散和危险度分层是影响患儿预后的独立危险因素(均P<0.05),见表2。
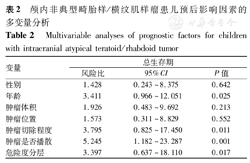
颅内非典型畸胎样/横纹肌样瘤患儿预后影响因素的多变量分析
Multivariable analyses of prognostic factors for children with intracranial atypical teratoid/rhabdoid tumor
颅内非典型畸胎样/横纹肌样瘤患儿预后影响因素的多变量分析
Multivariable analyses of prognostic factors for children with intracranial atypical teratoid/rhabdoid tumor
| 变量 | 总生存期 | ||
|---|---|---|---|
| 风险比 | 95%CI | P值 | |
| 性别 | 1.428 | 0.243~8.375 | 0.642 |
| 年龄 | 3.411 | 0.966~12.051 | 0.025 |
| 肿瘤体积 | 1.926 | 0.483~9.692 | 0.213 |
| 肿瘤位置 | 1.573 | 0.311~8.829 | 0.552 |
| 肿瘤切除程度 | 3.795 | 0.825~17.450 | 0.011 |
| 肿瘤是否播散 | 5.245 | 1.182~23.287 | 0.001 |
| 危险度分层 | 3.397 | 0.637~18.110 | 0.017 |
儿童颅内AT/RT是一种罕见病,肿瘤生长迅速,易复发,预后极差。本病男性多见,在本组病例中,男性患儿与女性患儿比例为4:1,高于以往报道[5]。幕下肿瘤比幕上肿瘤比例约为1.2:1.0,位于大脑半球脑实质比例不足30%,原发于脑室内的AT/RT非常罕见[6]。AT/RT的临床表现多样化,本组患儿多以颅内压增高症状起病,少数患儿因肿瘤位于大脑功能区(如基底核区、顶叶)以肢体活动障碍或癫痫起病。AT/RT影像学表现多样化,但并无特异性。结合本组患儿术前影像学表现,幕上AT/RT影像学表现可类似于儿童胶质瘤、原始神经外胚层肿瘤等,而幕下AT/RT有时难与髓母细胞瘤、室管膜瘤等相鉴别。总之,儿童颅内AT/RT初诊困难,确诊需依靠病理学诊断。AT/RT病理成分多种多样,通常含横纹肌样细胞及丰富的嗜酸性上皮样细胞,伴原始神经外胚层细胞(小圆形蓝细胞)及间充质细胞,常可见出血、坏死成分[7]。免疫组织化学有助于确诊AT/RT,INI1表达缺失是鉴别AT/RT的敏感和特异性方法,目前已广泛应用于临床[8]。随着分子病理的进展,分子基因检测也有助于AT/RT的确诊及分型,其特征主要为SMARCB1抑癌基因或SMARCA4抑癌基因的突变或缺失[9,10]。
由于AT/RT是一种罕见的神经系统肿瘤,且肿瘤生物学行为呈多样性,其治疗尚未形成统一的共识。本中心根据肿瘤的手术切除程度及肿瘤是否播散/转移对患儿进行了危险分层,并根据患儿年龄制定了个体化综合治疗方案。目前,多数学者认为最大范围的安全切除仍是首选治疗,大部分研究表明,肿瘤全切可延长患儿的生存期[11,12]。根据作者的经验,当肿瘤位于脑室时,部分患儿术前可合并梗阻性脑积水,此类患儿行脑室腹腔分流术应慎重,避免分流术造成肿瘤细胞的播散。另外,由于AT/RT瘤体血供常常极为丰富,术中应尽量沿肿瘤边界分离切除,避免过度的瘤内减压造成不可控制的出血。
多项研究表明,术后放疗可显著延长患儿的PFS期及OS期[7,13]。Yang等[14]最新的研究表明,术后先于全身化疗的早期放疗有助于改善患儿预后。全脑全脊髓放疗是术后辅助治疗的重要组成部分,但放疗的远期不良反应不容忽视,尤其在年龄较小的患儿。长期存活的患者可有认知功能下降、智力下降、生长发育迟缓等远期不良反应。Spina等[15]报道了应用立体定向放射外科(咖玛刀或射波刀)治疗的10例AT/RT患儿,66.7%的肿瘤治疗后体积减小或保持不变。Ren等[16]报道4例患儿接受联合咖玛刀的综合治疗后,获得了较为满意的效果。
目前的临床研究认为AT/RT对化疗敏感,化疗方案多为以铂类和烷化剂为基础[17]。由于AT/RT和髓母细胞瘤均为胚胎源性肿瘤,郑州大学第一附属医院基于髓母细胞瘤的化疗经验,临床上应用"CTX+DDP+VCR"化疗方案[18]。最近的一项研究表明,大剂量化疗联合鞘内注射MTX治疗可显著改善复发髓母细胞瘤患儿的预后[19]。且对于肿瘤未达全切和/或肿瘤播散/转移的患儿,给予鞘内注射治疗后,亦获得较为理想的治疗效果,患儿椎管内播散病灶可完全消失,脑脊液细胞学检查也可转阴。Thatikunta等[20]报道术前新辅助化疗可明显缩小肿瘤体积和减少供应血管密度,便于手术操作,提高肿瘤的全切除率。但作者认为,根据目前的诊断技术,仅基于影像学检查术前确诊AT/RT尚存在困难。未来,需要寻找AT/RT有效的诊断标志物,以提高术前诊断率,从而改进此类疾病的治疗方式。
由于儿童颅内AT/RT罕见,目前关于此类患儿预后相关因素的研究较少。已报道的有利预后因素包括肿瘤全切除、早期放疗及术后化疗,不利预后因素包括年龄<3岁、肿瘤播散、肿瘤转移等[16]。对本组患儿预后相关因素进行了分析,发现年龄≥3岁、肿瘤全切除、肿瘤未播散和标危组患儿预后较好。由于本组样本较小,且不同年龄段分组患儿术后放疗及化疗的方案不一,难以准确评价术后放疗及化疗对患儿预后的影响,未来需基于更大样本量进行研究评估。
由于AT/RT患儿预后极差,促使目前的治疗往更加激进的方向发展,包括尝试下探放疗的年龄,更大剂量的化疗方案结合自体干细胞移植等[14,21]。个体化的多学科联合治疗有助于延长患者的生存期。随着分子生物学的进展、精准医学的进步和基因分型的完善,AT/RT精准治疗将成为可能。对于不同危险度的患儿采用不同强度的治疗策略,将有助于进一步降低治疗的远期不良反应,提高生存率。期待未来基于多中心的研究,且在靶向治疗、免疫治疗等方面取得进展和突破,从而改善颅内AT/RT患儿的预后。
所有作者均声明不存在利益冲突

























