
细胞焦亡是一种新型的程序性细胞死亡,并伴有大量促炎症因子的释放。在心肌缺血再灌注损伤(MIRI)过程中,炎症反应贯穿着心肌损伤的整个病理生理过程。细胞焦亡由于具有高度促炎性,近年来研究者将其与MIRI紧密联系在一起。因此本文深入了解细胞焦亡的调控机制,以及对目前细胞焦亡在MIRI中的相关研究进行综述,为MIRI的临床防治研究提供一些思路。


随着社会老龄化比率的增长以及经济发展对居民饮食结构的改变,据2019年发布的中国心血管病报告显示,心血管疾病仍为我国病死率最高的疾病[1],特别是缺血性心肌病,病死率仅次于脑卒中。当心肌发生缺血时,尽早恢复组织血供被视为最重要的治疗手段,然而事实是恢复灌注后心肌的损伤变得更加严重,这种现象被称之为心肌缺血再灌注损伤(MIRI)。MIRI的发生除了在冠心病的急性梗阻后再通之外,还包括在心脏阻断下的外科手术、心肺复苏和心脏移植等情况中出现。因此关于MIRI的机制探讨和防治研究一直是心血管领域备受关注的话题。细胞焦亡是一种高度促炎性的细胞程序性死亡,由于其炎性特质而备受关注。近年来关于细胞焦亡介导MIRI的作用逐渐被报道。因此本文深入了解细胞焦亡的调控机制,以及对目前细胞焦亡在MIRI中的相关研究进行综述,为MIRI的临床防治研究提供一些思路。
1992年,Zychlinsky等首次研究发现福氏志贺氏菌诱导巨噬细胞发生的一种特异性的细胞凋亡。由于当时实验条件所限,因此未能详细阐述其特点。随后Chen等团队发现白细胞介素(IL)1β转化酶(ICE)这一重要蛋白,其在诱导这种特殊的细胞死亡起着关键作用。ICE属于半胱天冬酶(caspase)家族,由于第一个被发现,因此又被命名为caspase-1。早期研究者认为caspase-1主要负责切割IL-1β和IL-18发挥免疫调控作用。直到2001年,Cookson等提出以caspase-1介导并伴有高度促炎性新型细胞死亡,并命名为细胞焦亡。随着焦亡研究范围的扩大,人源性的caspase-4、5和鼠源性的caspase-11先后被发现。随后研究者统一将这些参与细胞焦亡发生的caspase统称为炎性caspase。关于炎性caspase是如何触发焦亡这一问题,将近长达二十多年的时间没有得到进一步的详细阐述。2015年,邵峰课题组详细地解答了这一问题,他们发现消皮素D(GSDMD)作为所有炎性caspase的共同底物,对诱导焦亡发生至关重要,是执行细胞焦亡发生的真正执行者[2]。
细胞焦亡本质是一种程序性细胞死亡,拥有部分凋亡的生物学特征,如核固缩、DNA断裂、Annexin V染色阳性等。但与凋亡又有着最本质的区别,如焦亡时细胞质膜的完整性出现缺失,并且伴有促炎因子释放。因此细胞焦亡被看作是一种兼具凋亡和坏死两种特征的新型细胞死亡形式。这种炎性程序性细胞死亡,又不同于坏死性凋亡,尽管二者质膜都形成小孔,但执行蛋白不同。坏死性凋亡执行蛋白是混合系列蛋白激酶样结构域(MLKL),其形成的小孔具有离子选择性,容易造成细胞内外形成渗透压差,导致细胞发生渗透性胀裂,而焦亡执行蛋白GSDMD在质膜上形成的小孔直径较大,无离子选择性,因而不容易发生渗透性胀裂[3]。关于细胞焦亡的发生机制目前以是否依赖caspase-1激活将其分为经典和非经典途径[2]。
1.经典途径:通过炎症小体模式识别受体(PRRs)识别细胞病原相关分子模式(PAMPs)或细胞死亡产生的损伤相关分子模式(DAMPs)并完成组装活化,活化的炎症小体可促进无活性的caspase-1前体分子(pro-caspase-1)裂解成为有活性的caspase-1,随后caspase-1进一步剪切炎症因子IL-1β和18前体(pro-IL-1β和pro-IL-18)以及GSDMD,启动细胞焦亡。其中炎性小体的激活被视为经典细胞焦亡途径的重要特征之一。
2.非经典途径:细胞依赖于caspase-4/5/11,可直接识别革兰阴性菌的脂多糖(LPS)并结合,随后通过剪切GSDMD启动焦亡进程。GSDMD作为炎性caspase的共同底物,其结构是由一个31 kDa的N端(GSDMD-N)结构域和一个22 kDa的C端(GSDMD-C)结构域构成。正常生理条件下GSDMD处于自抑状态,这主要源于GSDMD-C端通过连接环抑制了GSDMD-N端的细胞毒性。当炎性caspase对GSDMD中的连接环进行剪切后可解除这种自抑状态。解除抑制的GSDMD其N端可在质膜中发生寡聚形成小孔,从而破坏了细胞膜的完整性,造成细胞因子外漏[4]。GSDMD属于Gasdermin家族,目前研究报道的Gasdermin家族蛋白还有GSDMA、GSDMB、GSDMC、GSDME(DFNA5)及DFNB59等5种亚型[5]。近年来有研究报道,GSDME的N端与C端之间存在一个经典的caspase-3剪切位点,可以被caspase-3切割激活从而触发细胞焦亡[6]。由此,细胞焦亡的概念被重新定义为由Gasdermin介导的细胞程序性死亡。
MIRI是一个复杂且多因素综合的过程,如炎症反应、活性氧(ROS)生成、线粒体功能障碍、钙超载等,随着研究的深入发现这些因素并非独立存在而是相互交错最终导致心肌细胞发生不可逆的死亡。在MIRI过程中炎症始终贯穿着心肌损伤的整个病理生理过程,细胞焦亡由于具有高度促炎性,近年来研究者将其与MIRI紧密联系在一起。新近多项研究表明,细胞焦亡在MIRI的病理过程中发挥重要的作用,并且与上述MIRI存在机制交互。关于MIRI过程中触发的焦亡机制,目前主流观点是依赖于NLRP3/CASPASE-1/GSDMD这一重要信号途(图1)[7]。因此引出一种观点,即针对焦亡途径的成分调控可能是一种减少MIRI的方法。
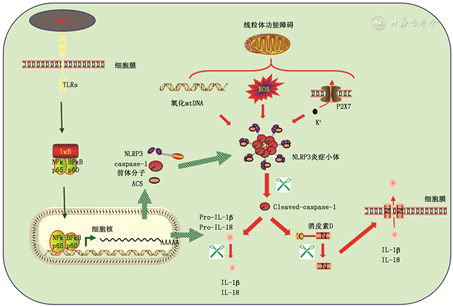
注:DAMPs为损伤相关分子模式;TLRs为Toll样受体;NLRP3为核苷酸寡聚化结构域样受体家族3;mtDNA为线粒体DNA;IL为白细胞介素;caspase为半胱天冬酶; ROS为活性氧;ASC为凋亡相关斑点样蛋白
1. NLRP3炎症小体与MIRI:炎症是机体的天然免疫系统防御机制。炎性小体的发现解释了机体是如何通过感受外界危险刺激并启动这种免疫保护机制。炎性小体是一个由多个蛋白组装而成的大分子复合体,其结构主要包括:PRRs、凋亡相关斑点样蛋白(ASC)和pro-caspase-1三个部分。PRRs通过识别PAMPs或DAMPs并向连接在caspase-1上的适配器ASC发出信号,促使未激活的pro-caspase-1聚集在一起。目前报道的PRRs有5种,包括:Toll样受体(TLRs)、Nod样受体(NLRs)、黑素瘤缺乏因子2样受体(ALRs)、维甲酸诱导基因Ⅰ样受体(RLRs)以及C型凝集素受体等家族。核苷酸寡聚化结构域样受体家族3(NLRP3)属于Nod样受体家族,因能感受多种刺激危险信号而被研究的最广泛,而目前关于NLRP3炎症体激活信号通路机制尚不完全清楚。
在正常心脏组织中NLRP3蛋白的表达丰度较低不足以激活炎症小体,因此在MIRI过程中,心肌组织NLRP3炎症小体的激活需要两步:第一步为启动阶段,主要是存活的心肌收到坏死或损伤的心肌释放的DAMPs刺激后,通过TLR-核因子κB(NF-κB)炎症信号转导途径,促进NLRP3、ASC、pro-caspase-1等蛋白表达并完成组装[8, 9]。第二步为触发阶段,完成组装的NLRP3炎症小体对再灌注后心肌的损伤机制作出应答反应,如ROS堆积、ATP诱导的K+外流、线粒体DNA(mtDNA)氧化等均可激活NLRP3炎症小体,随后通过促使caspase-1的活化而启动焦亡进程(图1)[10]。
Kawaguchi等[11]首次报道,在死于心肌梗死的患者中,心肌组织存有NLRP3炎症小体的组件ASC的凝聚。随后他们在小鼠的心肌梗死模型中发现,心肌成纤维细胞合成大量NLRP3 炎症小体并释放大量的IL-1β。为了证明NLRP3是MIRI的重要受体,Sandanger等[12]应用NLRP3敲除的成年小鼠,结果显示,与WT小鼠相比,NLRP3-/-小鼠缺血再灌注后心肌组织梗死面积减少,心功能明显改善。这些早期的研究预示着NLRP3炎症小体在MIRI的发展中的重要性。随后越来越多研究表明,心肌再灌注前使用药物抑制剂或shRNA基因干扰限制NLRP3炎症小体的活性均可改善心肌缺血再灌注后的损伤[13, 14]。为了详细阐述NLRP3炎症小体在MIRI中的调控机制。Liu等[15]研究显示,心肌I/R损伤后,硫氧还蛋白(Trx)-硫氧互作蛋白(TXNIP)与NLRP3相互作用增强NLRP3的表达。然而通过心肌内注射TXNIP siRNA降低NLRP3的活化可减少心肌梗死面积。但意外的是该研究发现在离体细胞实验中NLRP3在心肌微血管内皮细胞中内表达,而在心肌细胞中几乎不表达。因此作者认为TXNIP介导的心肌微血管内皮细胞NLRP3炎症小体激活是MIRI的一种新机制。Nazir等[16]研究表明,心肌缺血再灌注后,活化蛋白C(aPC)作为炎症的重要内源性抑制剂对心肌损伤具有保护作用,其保护机制与通过PAR-1和mTORC1抑制NLRP3的表达进而抑制caspase-1和IL-1β的激活有关。有意思的是Nazir等在体外实验中结果显示再灌注后的乳鼠心肌细胞NLRP3表达增高。这一结论与前面Liu等结果正好相反。目前关于再灌注后的心肌细胞中的NLRP3是否表达还存在争议。
2.caspase-1与MIRI:早期研究者认为caspase家族主要是介导细胞凋亡。随着一系列caspase同源序列的蛋白被不断地鉴定,caspases家族还参与介导多种不同的细胞死亡方式,其中包括细胞焦亡[17]。
多项实验研究显示,在小鼠心肌梗死模型中,通过基因技术敲除或限制小鼠的caspase-1的表达,可减少心肌梗死面积及降低左心室重构。因此caspase-1可作为防治心肌再灌注损伤的一个潜在的治疗靶点。VX-765是caspase-1的有效选择性抑制剂,它最初作为一种口服制剂用于治疗涉及caspase-1的疾病,如类风湿关节炎和癫痫。研究人员发现VX-765能够在MIRI动物模型中对caspase-1具有特定抑制作用。Yang等[18]首次在大鼠心肌梗死模型中发现在缺血前给予该药物对梗死具有有效的保护作用。随后Audia等[19]表明,再灌注时给予该药物也具有保护作用。该项研究者考虑到大多数经皮急性心肌梗死患者在再灌注前都使用了P2Y12血小板抑制剂,为了更好地模拟临床过程,研究者设置了VX-765联合P2Y12血小板抑制剂坎格雷洛(cangrelor)治疗组。结果显示VX-765联合cangrelor的治疗组的心肌梗死面积缩减比单独使用cangrelor或VX-765时要明显小,这表明这两种化合物在保护心脏方面没有共同的机制。为了观察VX-765对心肌组织的直接影响,作者又采用Langendorff灌注系统对大鼠心脏进行体外缺血再灌注建模,这样可以排除大鼠外周循环免疫细胞对心肌的保护作用,实验结果表明不仅VX-765在缺血开始时对离体心脏起到保护作用,其活性衍生物VRT-043198在再灌注开始时也同样具有保护作用。因此靶向caspase-1可作为MIRI防治的一种新的策略。
体外循环是心内直视手术必需的技术手段。尽管术中心肌保护措施及体外循环装置的不断改进,但MIRI仍是术后心功能障碍甚至导致死亡的主要原因。MIRI是一个复杂的过程,至今没有有效的治疗手段逆转心肌损伤,其具体机制目前仍不清楚。本课题组早期提出:急性心肌胰岛素抵抗(IR)是MIRI的又一重要机制[20, 21, 22],主要导致心肌细胞发生严重的能量代谢紊乱,而这些代谢异常不仅严重影响心脏功能,甚至导致心肌细胞死亡。NLRP3作为心肌细胞内重要的预警分子,可感受多种危险刺激信号,其中包括一些异常的代谢。由此推测在MIRI过程中,急性心肌IR可能是诱导心肌细胞焦亡的一重要始动因素,然而其具体作用途径仍有待探究。因此从心肌糖脂代谢紊乱的信号途径到进一步探索触发再灌注后心肌的炎性机制,为更全面地制定心肌保护策略有着特殊的理论指导作用和重要的临床参考价值。
所有作者均声明不存在利益冲突

























