
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
常染色体显性遗传性脑动脉病伴皮质下梗死和白质脑病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy,CADASIL)是一种遗传性全身性小血管疾病,由19号染色体Notch3基因突变引起,成年起病,临床表现为先兆偏头痛、情绪紊乱,反复短暂性脑缺血发作以及脑卒中,可导致严重残疾、痴呆和早亡[1]。Vogt-小柳原田(Vogt-Koyanagi-Harada,VKH)综合征又称葡萄膜脑膜脑炎,是一种以葡萄膜、脑膜、听觉系统和皮肤系统炎症为特征的自身免疫性疾病[2]。CADASIL合并VKH综合征病例少见报道,现将贵州省人民医院神经内科收治的1例患者诊治过程报道如下。
患者 女,42岁,因"左颜面部、左手麻木7 d"于2017年10月8日收治于我科。患者2016年开始出现反复头痛、精神抑郁、睡眠差、记忆力减退。否认高血压、糖尿病及烟酒嗜好。父亲有脑梗死病史。体检左面部针刺觉过敏。实验室检查结果提示:乙肝表面抗原、e抗体、核心抗体均阳性,乙型肝炎病毒-DNA定量1.91×104 IU/mL;抗核抗体-核颗粒型1∶100。头颅MRI示右侧丘脑急性期腔梗,脑白质多发异常信号,左侧基底节区、放射冠扩张血管间隙,双侧基底节区局灶性铁沉积(图1)。心电图、心脏B超、颈部血管B超、经颅多普勒超声均未见明显异常。拟做基因检测,未获同意。诊断:急性脑梗死(小动脉闭塞型),予抗血小板聚集、调脂稳斑、营养神经、改善循环等治疗后好转出院。
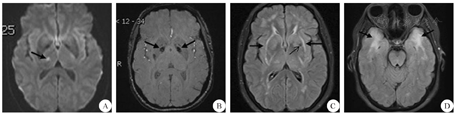
A:DWI上可见右侧丘脑新发梗死(箭头所示);B:SWI上可见双侧基底节区局灶性铁沉积(箭头所示);C:FLAIR上可见脑白质多发异常对称性高信号,累及外囊(粗箭头所示);左侧基底节区可见扩张血管间隙(细箭头所示);D:FLAIR上可见脑白质多发异常,累及双侧前颞叶(箭头所示);DWI:扩散加权成像;SWI:磁敏感加权成像;FLAIR:液体衰减反转恢复序列
2018年10月11日患者因"双眼视力下降6 d"再次入院,伴视物变形、视物变色,眼胀痛。病前2周有上呼吸道感染史。眼科体检:双眼视力指数/10 cm,红绿色不可辨,结膜轻充血,双侧瞳孔圆形等大,直径约4 mm,对光反射灵敏。神经系统体检:左侧Babinski征阳性,余无阳性体征。辅助检查:抗核抗体-核颗粒型1∶100。腰椎穿刺测脑压100 mmH2O(1 mmH2O=9.8 Pa),脑脊液外观无色透明,细胞总数48×106/L,有核细胞数28×106/L,蛋白0.59 g/L,氯、葡萄糖、腺苷脱氨酶正常,寡克隆区带IgG阴性、血清及脑脊液水通道蛋白4(AQP4)抗体阴性。脊髓MRI平扫及增强未见异常。眼底照相:视盘轻度水肿,血管走形迂曲(图2A)。眼底血管造影(FFA)示晚期视网膜可见环状荧光斑,相应视网膜荧光素积存,视盘荧光素着染;动脉变细(图2B)。光学相干断层扫描(OCT)显示,视网膜内见多灶性神经上皮层脱离,双眼视盘周围神经纤维层变厚(图2C)。纯音测听示左耳轻度、右耳轻微感音神经性听力下降。体感诱发电位及视觉诱发电位示多发颅内改变可能,双侧视觉、双上肢传导通路受累较重。头颅MRI示脑白质多发异常高信号,病变范围较前稍增多,右侧丘脑脑软化灶伴周围胶质增生,未见增强信号。基因检测示Notch3基因在Chr19:15302993位置上发生碱基C>T杂合变异,导致编码的153号氨基酸由精氨酸变异为半胱氨酸,Clinvar数据库收录该位点为致病性变异,经ACMG标准判断为可能致病(图3)。诊断VKH综合征、CADASIL,予甲基强的松龙1000 mg/d冲击治疗并序贯减量,联合维生素B1、维生素B12等治疗后患者双眼视力明显恢复。复查OCT示双眼病变较前减轻(图2D)。住院25 d,出院时仍有双眼视物时闪光,视物抖动,双眼视力均4.7。出院后予继续强的松、环磷酰胺、环孢素治疗。出院后3个月复查头颅MRI平扫及增强示脑白质多发异常高信号较前无明显变化,无强化信号。随访2年余,患者视力基本恢复正常,可正常工作,偶有头痛,无头昏、耳鸣,睡眠可,已停药1年。
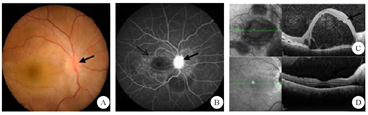
A:眼底照相,箭头示视盘轻度水肿;B:FFA,粗箭头示视盘荧光素着染,细箭头示视网膜环状荧光斑;C:OCT,箭头示视网膜神经上皮层脱离;D:复查时OCT;FFA:眼底血管造影;OCT:光学相干断层扫描
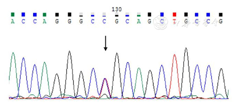
Notch3基因在Chr19:15302993位置上发生碱基C>T杂合变异
本例患者有脑卒中家族史,以脑梗死为主要表现,伴偏头痛、情绪障碍及记忆力减退,颅脑MRI见丘脑腔隙性梗死、双侧大脑半球对称性和进行性的白质病变,累及前颞叶和外囊,伴有多发陈旧腔隙性梗死灶、扩张的血管周围间隙、脑微出血,符合CADASIL颅脑MRI表现,基因检测示Notch3基因致病性突变,CADASIL诊断明确。患者后来出现急性双眼视力下降,检查见双眼弥漫性脉络膜视网膜炎、视盘水肿、渗出性视网膜脱离,符合VKH综合征眼部表现。脑脊液蛋白及细胞轻度增高、双耳轻度听力下降,提示存在脑膜炎、听力受损等VKH综合征眼外表现。患者无脊髓病变,AQP4抗体阴性,可除外视神经脊髓炎谱系疾病;头颅MRI示病灶累及双侧颞叶,伴情绪障碍及记忆力减退,需与自身免疫性脑炎鉴别,但病程中多次头颅MRI示脑白质多发异常信号均无明显改变,不符合自身免疫性脑炎治疗后的影像表现,故排除。
CADASIL和VKH综合征都可波及到中枢神经系统和眼部,二者临床表现及影像学表现既有交叉又各有特点。CADASIL可累及眼底小血管,常表现为前部缺血性视神经病变、眼底动脉硬化狭窄、视网膜神经纤维层缺失等病变[3];而VKH综合征眼底表现为双眼弥漫性脉络膜视网膜炎、视盘水肿、渗出性视网膜脱离[2];二者眼部损害容易鉴别。本例患者FFA检查发现除了有典型VKH综合征的表现,同时见眼底小动脉变细硬化,提示可能有CADASIL眼底小血管的损伤。VKH综合征是一种自身免疫性肉芽肿疾病,由CD4+T细胞介导,靶向黑素细胞抗原,这些抗原广泛存在于眼、中枢神经系统、内耳和皮肤[4]。中枢神经系统髓鞘碱性蛋白是一种酪氨酸相关蛋白,可能会受到VKH综合征相关免疫损伤的攻击,导致脑白质病变,一般表现为脑室周围或脑干小片状或双侧对称性弥漫白质病变,但无CADASIL特征性的双侧外囊及前颞叶受累表现[5,6]。
CADASIL与自身免疫病共患罕见报道,但并非偶然。Paraskevas等[7]报道的1个家系中有3例CADASIL患者发生Notch3 p.R169C突变,同时合并系统性自身免疫性疾病,包括抗磷脂综合征、自身免疫性血小板减少和IgA肾病。CADASIL是由Notch3基因突变引起的遗传性小血管疾病。Notch3基因突变使Notch3受体半胱氨酸残基增加或减少,产生未能配对的半胱氨酸残基,导致Notch3受体胞外结构域在小血管中过度聚集,促进细胞外基质蛋白异常募集,使小血管平滑肌细胞变性[8]。Notch信号通路介导细胞间通信,控制细胞命运走向,通过调节免疫细胞的分化、增殖以及调控细胞因子的水平,在免疫疾病发病机制中发挥作用[9]。巨噬细胞迁移抑制因子(migration inhibitory factor,MIF)是重要的促炎细胞因子,MIF过表达可激活Notch信号通路,使Notch1和Notch4表达上调,引起辅助性T细胞(Th)1和Th17细胞数量及活性增加,加重实验性自身免疫性葡萄膜炎小鼠眼部炎症[10]。研究发现葡萄膜炎发病过程中伴有Notch1、白细胞介素(IL)-10、IL-17表达升高以及CD4+/CD8+、Th17/Treg比值失衡[11]。活动期VKH综合征患者CD4+T细胞中Notch1、Notch2以及Notch3 mRNA表达及蛋白水平较静止期和健康对照者显著升高,抑制Notch信号通路可降低活动期VKH综合征患者CD4+T细胞分泌IL-22水平,提示Notch信号通路可通过调控CD4+T细胞分泌IL-22参与VKH综合征的发病[12]。
综上,我们推测,血管和免疫系统之间可能通过Notch信号通路发生相互联系。CADASIL患者突变的Notch3受体胞外结构域在血管平滑肌异常聚集,可能作为一种抗原表位,激活Notch信号通路免疫反应,诱导Notch3受体蛋白下游信号分子活化,参与葡萄膜炎症损伤,导致VKH综合征发生。这种假设可能是本例CADASIL合并VKH综合征的机制之一。不足的是,本例患者未检测Notch3、T淋巴细胞亚群以及IL-22等细胞因子的表达水平,这种假设尚需研究证实。此外,本例患者感染乙型肝炎病毒,可引起多种特异性T淋巴细胞反应,可导致肝细胞损伤[13]。这种特异性T细胞反应是否参与本例VKH综合征患者的发病尚需要进一步研究。
所有作者均声明不存在利益冲突

























