
探讨遗传性凝血因子FⅪ缺乏症患者的基因突变、临床特点,以及诊断与治疗方法。
选择2020年8月10日及2020年10月30日,四川大学华西医院血液科收治的2例遗传性FⅪ缺乏症患者为研究对象。按照就诊时间顺序,将2例患者分别编号为患者1和2。根据2例患者的凝血功能指标检查,以及F11基因测序结果等,对患者进行诊断;根据其临床出血症状和程度,给予临床观察或者对症治疗。通过对人类基因突变数据库(HGMD),单核苷酸多态性数据库(dbSNP)及PubMed数据库进行检索,确认基因测序分析检出基因突变是否为新发现基因突变。采用Mutation Taster、Polyphen-2及PROVEAN在线软件,对新发现的错义突变位点进行致病性预测。对患者的随访截至2021年2月28日。采用回顾性分析方法,对2例患者的临床表现与诊治过程进行分析。检索中国知网数据库、万方数据服务知识平台、PubMed数据库中与本研究患者F11基因突变相同或者相似的病例报道文献。文献检索时间为数据库建库至2021年2月28日。总结与本研究遗传性FⅪ缺乏症患者相关的基因突变类型及出血表现。本研究符合2013年修订的《世界医学协会赫尔辛基宣言》要求,并且获得患者知情同意,与患者签订临床研究知情同意书。
①病史采集:患者1,男性,24岁,因"左大腿软组织外伤后血肿,术前检查发现凝血功能异常"就诊。患者自诉无明显不适。患者2,女性,32岁,因"孕前体检发现活化部分凝血活酶时间(APTT)延长"就诊。患者诉平素偶有皮肤淤斑。②实验室检查结果:患者1的APTT、凝血酶原时间(PT)、FⅪ活性(FⅪ∶C)分别为97.9 s、11.9 s、0.6%;患者2的上述3项指标分别为95.4 s、12.5 s和1.2%。③基因测序:患者1伴F11基因复合杂合突变,包括杂合错义突变c.149G>T(p.Cys50Phe)与杂合无义突变c.1204C>T(p.Gln402Ter);患者2伴F11基因纯合缺失突变c.1058delA(p.Asn353ThrfsTer18)。④F11基因错义突变c.149G>T(p.Cys50Phe)及缺失突变c.1058delA(p.Asn353ThrfsTer18)均为新突变,并且具有致病性。⑤治疗及随访结果:2例患者均仅接受临床观察,截至随访结束,患者一般情况良好。⑥文献复习结果:筛选出的6篇相关文献报道的10例该病患者中,发生F11基因杂合无义突变c.1204C>T(p.Gln402Ter)及该突变相邻位置无义突变c.1202C>T(p.Trp401Ter)、错义突变c.149G>T(p.Cys50Phe)邻近位置的错义突变致病者分别为1、3、6例。
本研究发现遗传性FⅪ缺乏症患者2种F11基因新突变,包括错义突变c.149G>T(p.Cys50Phe)及缺失突变c.1058delA(p.Asn353ThrfsTer18),均导致患者FⅪ活性降低。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
遗传性凝血因子FⅪ缺乏症是由F11基因突变所致的罕见出血性疾病(rare bleeding disorders, RBD)。该疾病在人群中的发病率约为1/100万[1]。遗传性FⅪ缺乏症在世界血友病联盟(World Federation of Hemophilia,WHF)和罕见出血性疾病欧洲网络(European Network of Rare Bleeding Disorders, EN-RBD)资料库登记的RBD病例中最常见,约占1/3[2]。遗传性FⅪ缺乏症为常染色体隐性遗传病,也有文献报道部分F11基因错义突变(p.Ser225Phe和p.Cys398Tyr)编码的错误FⅪ蛋白通过与野生型FⅪ形成异源二聚体,影响FⅪ蛋白分泌,从而导致部分病例呈显性遗传表型[3,4]。遗传性FⅪ缺乏症患者的临床症状通常表现为轻、中度出血,出血严重程度与血浆中FⅪ活性(FⅪ∶C)及基因突变类型无明显相关性,而与出血部位相关[2,5,6]。本研究对四川大学华西医院确诊的2例遗传性FⅪ缺乏症患者进行基因测序,确证了2种新的遗传性FⅪ缺乏症基因突变,进一步对这2例患者的临床资料进行分析,并且对与本研究2例患者F11基因突变相同或者突变位点邻近的遗传性FⅪ缺乏症的研究文献进行复习,旨在提高临床医师对该病临床特点及分子机制的认识。现将研究结果报道如下。
选择2020年8月10日及2020年10月30日,于四川大学华西医院血液科收治的2例遗传性FⅪ缺乏症患者为研究对象。按照就诊时间顺序,将2例患者分别编号为患者1和2。本研究符合2013年修订的《世界医学协会赫尔辛基宣言》要求,并且获得患者知情同意,与患者签订临床研究知情同意书。
目前临床对于遗传性FⅪ缺乏症尚无确切的诊断与治疗标准,诊断主要依据患者的临床表现、FⅪ∶C及F11基因突变分析结果[7,8]。治疗主要依据患者的临床出血症状和程度,给予临床观察或者对症治疗[7,8]。
分别于2020年8月10日及2020年10月30日,使用0.109 mmol/L枸橼酸钠抗凝管,采集2例患者外周静脉血7 mL。取2 mL枸橼酸钠抗凝全血标本用于实验室检查,检测项目包括:活化部分凝血活酶时间(activated partial thromboplastin time,APTT),凝血酶原时间(prothrombin time,PT),APTT纠正试验,FⅪ∶C,FⅪ血浆抗体检查,凝血酶时间(thrombin time,TT),国际标准化比值(international normalized ratio,INR),以及肝、肾功能检查等。
取5 mL枸橼酸钠抗凝全血标本,采用DNA提取试剂盒提取患者基因组DNA。采用Primer Blast(https://www.ncbi.nlm.nih.gov/blast.cgi)设计覆盖F11基因所有编码区外显子及侧翼序列的引物共13对(表1),并送至擎科生物科技有限公司合成。对2例患者DNA进行PCR扩增。将PCR扩增产物送至四川华西康圣达血液病特检中心采用Sanger测序法进行F11基因正、反向测序分析,测序结果采用Mutation Surveyor v5.1.1软件(美国Soft Genetics公司)与美国国家生物技术信息中心(National Center for Biotechonlogy Information,NCBI)基因库公布的参考序列进行对比,寻找突变位点。

F11基因PCR扩增引物
F11基因PCR扩增引物
| 外显子 | 引物序列 | 扩增产物片段大小(bp) | |
|---|---|---|---|
| Exon2 | 190 | ||
| 正向引物 | GTAAAACGACGGCCAGTGCCCCTCTCTCCCTATTTCTTGTA | ||
| 反向引物 | GGAAACAGCTATGACCATGCTCTCCCAGCCCATAGTTAAGAT | ||
| Exon3 | 535 | ||
| 正向引物 | GTAAAACGACGGCCAGTGCCAACGTACCATTTTGTGATTTT | ||
| 反向引物 | GGAAACAGCTATGACCATGATTGCGTAAACACCATCACTTCT | ||
| Exon4 | 478 | ||
| 正向引物 | GTAAAACGACGGCCAGTGACACAGATTTTTGGTTGCATTTT | ||
| 反向引物 | GGAAACAGCTATGACCATGCCACATAGCAGTGTTTCATTTCA | ||
| Exon5 | 524 | ||
| 正向引物 | GTAAAACGACGGCCAGTGCATTGGCATTCTTTTACTGCTTC | ||
| 反向引物 | GGAAACAGCTATGACCATGCCATAAATGAAATGCATCCAAAT | ||
| Exon6 | 420 | ||
| 正向引物 | GTAAAACGACGGCCAGTGGGTCACTCTCTCAAGCTGTCATT | ||
| 反向引物 | GGAAACAGCTATGACCATGAGCATAAGCTGGTATCCTGAGTG | ||
| Exon7 | 336 | ||
| 正向引物 | GTAAAACGACGGCCAGTGGGTGAATTGAGTCCCTGACATAG | ||
| 反向引物 | GGAAACAGCTATGACCATGGGGCTAGAAATTCTTTGCAACTT | ||
| Exon8 | 582 | ||
| 正向引物 | GTAAAACGACGGCCAGTGATAAACCTATTGCAACCCTGGAT | ||
| 反向引物 | GGAAACAGCTATGACCATGCTAGACAGCAGACCACATGTCAG | ||
| Exon9-10 | 557 | ||
| 正向引物 | GTAAAACGACGGCCAGTGAAACTGAGAGTTCTGCATTCTGG | ||
| 反向引物 | GGAAACAGCTATGACCATGTGTTCTCCCTTCTGTGGCTATAA | ||
| Exon11 | 516 | ||
| 正向引物 | GTAAAACGACGGCCAGTGAGTCAATTCCATTTTTCATGTGC | ||
| 反向引物 | GGAAACAGCTATGACCATGGAGACCATGAGCTCTTCCTTTTT | ||
| Exon12 | 393 | ||
| 正向引物 | GTAAAACGACGGCCAGTGAGGAAATTTCTTTCCCTCTGTTG | ||
| 反向引物 | GGAAACAGCTATGACCATGGTTTCAATAGCCAGGAAAGTGTG | ||
| Exon13 | 280 | ||
| 正向引物 | GTAAAACGACGGCCAGTGGAGGAAAATACACGACAACAAGG | ||
| 反向引物 | GGAAACAGCTATGACCATGTTCATTTCTAGGATGGAGCACAT | ||
| Exon14 | 204 | ||
| 正向引物 | GTAAAACGACGGCCAGTGTGGTTATTCTACAAACGAACCAAA | ||
| 反向引物 | GGAAACAGCTATGACCATGTCCATTGGCTAAGAACACTCTGT | ||
| Exon15 | 406 | ||
| 正向引物 | GTAAAACGACGGCCAGTGATATTAAGGGGCCAAGACAACAT | ||
| 反向引物 | GGAAACAGCTATGACCATGGCATTTTCTTACAAACACCAAGC | ||
通过对人类基因突变数据库(Human Gene Mutation Database, HGMD, http://www.hgmd.cf.ac.uk),单核苷酸多态性数据库(Single Nucleotide Polymorphism Database, dbSNP)及PubMed数据库进行检索,确认基因测序分析所得基因突变是否为新基因突变。采用Mutation Taster (http://www.mutationtaster.org/), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/)及PROVEAN (http://provean.jcvi.org/index.php)软件对新发现的错义突变位点进行致病性预测。
本研究随访截至2021年2月28日。随访期间通过门诊或者电话随访方式收集患者相关临床表现及出血症状。
本研究以"遗传性FⅪ缺乏""先天性FⅪ缺乏""血友病C""congenital FⅪ deficiency""inherited FⅪ deficiency"为中、英文关键词,在中国知网数据库、万方数据服务知识平台、PubMed数据库中,检索有关遗传性FⅪ缺乏症的相关研究文献,通过阅读文献题目及摘要排除无关文献。通过阅读全文筛选出与本研究2例患者F11基因突变相同或者突变位点邻近的遗传性FⅪ缺乏症的研究文献。检索时间为数据库建库至2021年2月28日。
采用回顾性分析方法,对2例遗传性FⅪ缺乏症患者的临床病例资料进行分析。同时,根据本研究设定的检索策略对检索所得遗传性FⅪ缺乏症相关文献进行复习,并对遗传性FⅪ缺乏症患者的临床表现及基因突变进行分析、总结。
患者1,男性,24岁,因"左大腿软组织外伤后血肿,术前检查发现凝血功能异常"于2020年8月10日就诊于本院血液科。患者未诉明显不适,左大腿手术伤口愈合良好,无口腔、牙龈及鼻出血,无皮下淤点、淤斑。患者既往体健,家系中一、二级亲属均无出血表现。患者2,女性,32岁,因"孕前体检发现APTT延长"于2020年10月30日就诊于本院血液科。患者诉平素偶有皮肤淤斑,无口腔、牙龈及鼻出血。患者既往体健,家系中一、二级亲属均无出血表现。
相关实验室检查结果示,患者1的APTT为97.9 s,PT为11.9 s,APTT纠正试验结果正常,FⅪ∶C为0.6%,FⅪ血浆抗体检查结果呈阴性,其他凝血指标正常,肝、肾功能正常。患者2的APTT为95.4 s,PT为12.5 s,APTT纠正试验结果正常,FⅪ∶C为1.2%,其他凝血指标正常,肝、肾功能正常。2例遗传性FⅪ缺乏症患者一般临床资料,见表2。
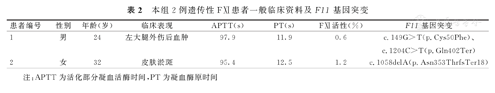
本组2例遗传性FⅪ患者一般临床资料及F11基因突变
本组2例遗传性FⅪ患者一般临床资料及F11基因突变
| 患者编号 | 性别 | 年龄(岁) | 临床表现 | APTT(s) | PT(s) | FⅪ活性(%) | F11基因突变 |
|---|---|---|---|---|---|---|---|
| 1 | 男 | 24 | 左大腿外伤后血肿 | 97.9 | 11.9 | 0.6 | c.149G>T(p.Cys50Phe)、c.1204C>T(p.Gln402Ter) |
| 2 | 女 | 32 | 皮肤淤斑 | 95.4 | 12.5 | 1.2 | c.1058delA(p.Asn353ThrfsTer18) |
注:APTT为活化部分凝血活酶时间,PT为凝血酶原时间
基因测序结果显示,患者1伴F11基因复合杂合突变,包括位于3号外显子杂合错义突变c.149G>T与位于11号外显子杂合无义突变c.1204C>T,分别导致3号外显子第50位半胱氨酸突变为苯丙氨酸(p.Cys50Phe),以及11号外显子第402位编码谷氨酰胺的密码子突变为终止密码子(p.Gln402Ter)。患者2伴F11基因10号外显子纯合缺失突变c.1058delA,导致移码突变,使353位氨基酸后的序列发生改变,并在其后第18位提前产生终止密码子(p.Asn353ThrfsTer18)。本组2例患者基因测序结果,见图1,图2。

注:蓝色方框代表突变位点
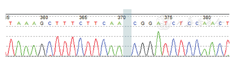
注:蓝色方框代表突变位点
经检索HGMD、dbSNP及PubMed数据库确认F11基因c.149G>T(p.Cys50Phe)及c.1058delA(p.Asn353ThrfsTer18)突变为国内、外尚未见报告的新突变。c.1058delA (p.Asn353ThrfsTer18)突变为终止密码,产生截短的无功能FⅪ蛋白,因此确定其致病性。本研究对错义突变c.149G>T(p.Cys50Phe)进行致病性预测结果显示,Mutation Taster预测其为致病性突变,Polyphen-2评分为1分,预测其为有害突变,PROVEAN评分为-5.331分,预测其为有害突变。
本组2例患者FⅪ∶C减低,但是患者及家庭成员既往均无明显出血表现,因此未予血浆输注等特殊治疗,仅临床观察随访。截至随访结束,2例患者一般情况良好,无鼻出血、口腔牙龈等出血情况。
根据本文设定的文献检索策略进行检索的结果显示,共计筛选出6篇国内、外与错义突变c.149G>T(p.Cys50Phe)及无义突变c.1204C>T(p.Gln402Ter)相同或位置邻近的遗传性FⅪ缺乏症研究的相关文献[9,10,11,12,13,14]。其中1例患者由杂合无义突变c.1204C>T(p.Gln402Ter)引起,3例患者由该突变相邻位置无义突变c.1202C>T (p.Trp401Ter)引起;未检索出由杂合错义突变c.149G>T(p.Cys50Phe)引起的遗传性FⅪ缺乏症的患者,但有6例患者由该突变邻近位置的错义突变致病。此外,未检索出伴与缺失突变c.1058delA(p.Asn353ThrfsTer18)相同或者邻近位置突变导致的遗传性FⅪ缺乏症的文献报道。本研究文献复习结果,见表3。

伴与本研究F11基因突变位点相同或者邻近位点突变的遗传性FⅪ缺乏症病例报道
伴与本研究F11基因突变位点相同或者邻近位点突变的遗传性FⅪ缺乏症病例报道
| 文献(第一作者,文献发表年) | 例数 | F11基因突变 | 伴出血症状者例数 |
|---|---|---|---|
| Fard-Esfahani等[9],2008 | 2 | 患者1:杂合错义突变c.151A>C(p.Thr51Pro)患者2:杂合错义突变c.152C>T(p.Thr51Ile)、 | 1(患者1) |
| Bicocchi等[10],2011 | 2 | 杂合错义突变c.140A>C(p.Gln47Pro) | 0 |
| Mitchell等[11],2006 | 1 | 杂合错义突变c.141G>C(p.Gln47His) | 0 |
| Kim等[12],2012 | 1 | 杂合错义突变c.159C>A(p.His53Gln) | 1 |
| 武文漫等[13],2003 | 3 | 杂合错义突变c.1202C>T(p.Trp401Ter) | 0 |
| Shao等[14],2016 | 1 | 纯合错义突变c.1204C>T(p.Gln402Ter) | 0 |
FⅪ是由肝细胞和巨核细胞合成的丝氨酸蛋白酶原,以同源二聚体的形式存在于外周血循环,每个单体包含4个apple结构域(apple domain,AP) 1~4和1个丝氨酸蛋白酶催化结构域(serine protease domain,SP)[15]。每个AP由7个反向平行β折叠和其包绕的1个α螺旋组成,其内部的二硫键起稳定结构域的作用。AP1~3分别与凝血酶、高分子激肽酶原、血小板、肝素及FⅪ相互作用,AP4上的321Cys形成二硫键参与二聚体形成[16,17]。在体内,FⅪ的主要激活物为凝血酶,活化的FⅪ通过激活FⅨ参与凝血级联反应。此外,FⅪ有助于凝血酶激活的纤溶抑制物活化,因此FⅪ缺乏症患者在局部纤溶活跃的部位更易发生出血,如口腔黏膜、泌尿道等[6]。
F11基因位于4号染色体4q35,包含15个外显子及14个内含子,1号外显子编码5′-非翻译区,2号外显子编码信号肽,3~10号外显子分别编码4个AP,外显子11~15编码催化结构域[18,19]。目前,HGMD已收录F11基因突变270余种,其中最主要的突变为错义突变和无义突变,其他突变还包括插入、缺失及剪切突变,突变分布于F11基因的各个区域,相对集中于编码催化结构域的区域。
本研究中,患者1临床表现为外伤后血肿,患者2表现为偶发皮肤淤斑,平素均无其他出血表现。2例患者实验室检查结果均显示APTT延长,PT正常,符合FⅪ缺乏症轻、中度出血的临床特征。文献报道,遗传性FⅪ缺乏症是孕期女性患者流产的危险因素[20],因此患者妊娠后,应密切观察随访。基因测序结果显示,患者1伴F11基因复合杂合突变,其中一处位于11号外显子区域(c.1204C>T),为杂合无义突变,导致第402位编码谷氨酰胺的密码子突变为终止密码子,产生一个截短的蛋白,失去催化结构域。该突变首先在一个中国家系中被报道,该患者为纯合突变,仅伴轻微出血表现[14]。此外,文献报道,在F11基因无义突变p.Gln402Ter相邻位置也存在杂合无义突变c.1202C>T(p.Trp401Ter),引起遗传性FⅪ缺乏症,但是患者均无出血表现[13]。本研究发现,F11基因3号外显子区域杂合错义突变c.149G>T,导致第50位编码半胱氨酸的密码子转变为编码苯丙氨酸的密码子。经过数据库检索和文献查阅,该突变为新突变,3个遗传变异在线分析软件(Mutation Taster、Polyphen-2及PROVEAN)均预测其为致病突变。p.Cys50Phe位于AP1结构域,该位点半胱氨酸突变成苯丙氨酸导致该处二硫键无法形成,因此可能影响AP1的结构稳定性,从而导致FⅪ缺乏。也有文献报道,在F11基因错义突变p.Cys50Phe相邻位置的氨基酸改变(p.Gln47Pro、p.Gln47His、p.Thr51Pro、p.Thr51Ile、p.His53Gln)引起的遗传性FⅪ缺乏症,除p.Thr51Pro引起患者术中出血外,其余突变患者均无明显出血倾向[9,10,11,12]。遗传性FⅪ缺乏症为常染色体隐性遗传病。文献报道,某些错义突变通过与野生型FⅪ形成异源二聚体发挥显性负效应,使得部分病例呈显性遗传的表型[3,4]。该患者发生3号和11号外显子的复合杂合突变,这种复合的杂合突变可能共同引起FⅪ的活性降低,导致患者临床发病。患者2检测出位于10号外显子的纯合缺失突变c.1058delA,此突变导致氨基酸序列从353位开始发生改变,并且在其后第18位变为终止密码子,提前终止翻译,产生一个截短的蛋白,从而丢失AP4及SP,蛋白结构的完整性受到影响,导致FⅪ活性降低而发病。经检索HGMD、dbSNP及PubMed数据库发现,F11基因错义突变c.149G>T (p.Cys50Phe)及缺失突变c.1058delA (p.Asn353ThrfsTer18)目前尚未见文献报道。
本研究对2例遗传性FⅪ缺乏症患者进行了基因突变分析,发现2种国内、外尚未报道的F11基因突变,分别为错义突变c.149G>T(p.Cys50Phe)及缺失突变c.1058delA(p.Asn353ThrfsTer18)。2种突变均与患者的FⅪ活性降低相关。本研究结果完善了遗传性FⅪ缺乏症基因突变种类。
所有作者均声明不存在利益冲突

























