
对2017年3月复旦大学附属儿科医院收治1例以胆管扩张为重要表现的CD40LG新发基因突变X连锁高IgM综合征(XHIGM)患儿的临床资料进行回顾性分析。患儿为4岁男童,因反复腹泻0.5年、皮肤黄染5 d入院。胎儿期及出生史无异常;婴儿期曾患2次重症肺炎,左腋下淋巴结化脓性感染。体检发现体格发育明显落后,重度营养不良。皮肤、巩膜中度黄染,浅表淋巴结大,肝大。血白细胞、嗜酸性粒细胞升高、C反应蛋白明显升高;血红蛋白、白蛋白低下,高γ-谷氨酰转肽酶性胆汁淤积,免疫球蛋白IgG低下,IgM正常。影像学检查提示肝内外胆管弥散性扩张;肝脏病理发现胆小管增生,部分大胆管周围纤维组织增生;基因检测高通量测序发现XHIGM基因CD40LG致病性突变(exon5 c.506A>G,p.Y169C),母亲为携带者。入院后给予抗感染、调整饮食、白蛋白、静脉丙种球蛋白支持治疗,熊去氧胆酸利胆治疗,患儿病情好转出院。该病例提示XHIGM除常见的感染特点外,也可表现为弥散性肝内外胆管扩张、嗜酸性粒细胞明显升高。CD40LG基因exon5 c.506A>G是该病的致病性突变。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
高IgM综合征(HIGM)是一种罕见的原发性免疫缺陷病,其主要特点为外周血IgM水平正常或增高,伴IgG、IgA和IgE水平降低,而B淋巴细胞数正常[1]。X染色体上的CD40配体(CD40L)基因缺陷是HIGM的最常见原因,称为X连锁高IgM综合征(XHIGM),占HIGM的65%~70%[2],CD40LG是导致XHIGM的基因。自1960年HIGM首次被报道后,国内外均有病例陆续报道,其主要临床表现为反复呼吸道感染、慢性腹泻、中性粒细胞减少症、胆管炎等[3],查阅文献未见以肝内外胆管扩张为重要表现的XHIGM。由于患者的这些非典型免疫球蛋白谱和表型,使得识别比较困难,常延误诊断,现报道1例以肝内外胆管扩张为重要表现的CD40LG基因突变致XHIGM,分享一些临床诊疗经验并改善上述情况。
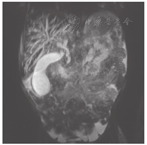
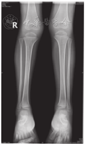
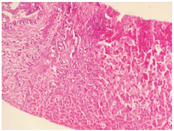
患儿,男,4岁,因"间断腹泻0.5年,皮肤、巩膜黄染5 d"于2017年3月11日就诊于复旦大学附属儿科医院。患儿0.5年前开始反复出现油腻饮食后腹泻,为稀糊便,1~2次/d,无黏液脓血,无发热、腹痛、皮肤黄染,可自行缓解。曾予中成药调理,无明显改善。入院前5 d因体检发现贫血后口服铁剂治疗,渐出现皮肤、巩膜黄染,无发热,不伴腹痛、腹泻及大便颜色变浅,无皮肤瘙痒,无纳差、乏力、恶心、呕吐,小便深黄。发病后曾到当地医院住院治疗2 d,检查发现肝功能异常,贫血、白蛋白低下、凝血功能异常、IgG低下,为进一步诊治,转至复旦大学附属儿科医院。患儿系第1胎,第1产,足月顺产,出生体质量3 650 g,无窒息及抢救史。患儿出生后母乳喂养,按时添加辅食。运动及智力发育与同龄儿相仿;出生后接种卡疫苗后出现左腋下淋巴结脓肿;婴儿期曾2次患重症肺炎并行机械通气。父母体健,非近亲结婚,无明确家族性、遗传性疾病史。体格检查:体质量13 kg,身高95 cm。贫血貌。颈部及腋下可触及黄豆大小淋巴结,皮肤及巩膜中度黄染,无肝掌、蜘蛛痣。心肺查体无特殊,肝脏肋下3 cm,剑突下3 cm,质韧,脾脏肋下未触及。主要实验室检查:血常规白细胞18.4×109/L,嗜酸性粒细胞0.188,血红蛋白84.2 g/L,中性粒细胞0.105,血小板557×109/L,网织红细胞0.064,C反应蛋白64 mg/L;血生化:白蛋白23.9 g/L,碱性磷酸酶171 IU/L,丙氨酸转氨酶132 IU/L,天冬氨酸转氨酶199 IU/L,磷酸肌酸激酶1 710 IU/L,肌酸激酶105 U/L,肌酸激酶同工酶105 IU/L,结合胆红素93.2 μmol/L,γ-谷氨酰转移酶673 IU/L,球蛋白14.7 g/L;钾4.3 mmol/L,乳酸脱氢酶571 IU/L,血氨78 μmol/L;Coomb′s试验阴性;血甲胎蛋白、癌胚抗原、铁蛋白、神经元特异性烯醇化酶均正常;血IgA 0.44 g/L,IgG 2.40 g/L,IgM 0.97 g/L;总IgE 10.0 kU/L;CH50正常,补体C3、C4正常;流式细胞检测CD系列未见明显异常,活化淋巴细胞正常;结核菌T细胞斑点检测(T-SPOT)阴性,血寄生虫抗体阴性,大便常规、隐血及寄生虫检查均未见异常;支气管镜肺泡灌洗液:抗酸杆菌阴性,灌洗液半乳甘露聚糖(GM)试验阴性;腹部磁共振成像(MRI)、磁共振胰胆管成像(MRCP)示肝内胆管、肝总管、胆囊及胆总管扩张(图1);胸部CT示两肺多发斑片、条索状高密度影,局部与邻近胸膜黏连,局部支气管管腔扩张。胸腔见少量积液。骨X线片:双下肢诸骨骨质稀疏(图2);肝脏病理:肝细胞肿胀,部分气球样变,部分肝细胞胞质内见小空泡形成,少量肝细胞内胆汁淤积;部分肝血窦轻度扩张,枯否细胞轻度增生;汇管区纤维组织增生,胆小管增生,部分大胆管周围纤维组织增生,较多嗜酸性粒细胞及少量淋巴细胞、中性粒细胞浸润(图3)。胃镜及小肠胶囊内镜提示浅表性胃炎(轻度),小肠黏膜炎;胃窦黏膜病理活检:轻度慢性炎症,幽门螺杆菌(-)。骨髓涂片未见明显异常。入院后积极给予无脂且蛋白以植物蛋白为主饮食,头孢曲松抗感染,静脉丙种球蛋白、白蛋白、补充脂溶性维生素支持治疗,熊去氧胆酸利胆,患儿腹泻、胆汁淤积好转。获得患儿监护人知情同意后对患儿家系进行高通量测序发现,患儿CD40LG基因突变(exon5 c.506A>G,p.Y169C),169位氨基酸有酪氨酸变为半胱氨酸,为致病性突变(rs786205606),母亲为携带者,为已知X连锁隐性遗传性疾病致病性基因突变。
本研究获得复旦大学附属儿科医院医学伦理委员会批准[批准文号:复儿伦审(178号)],患儿监护人均知情同意。
CD40L为肿瘤坏死因子(TNF)超家族成员,属Ⅱ类跨膜蛋白,由CD40LG编码,其基因定位于X染色体的q26.32q27.1区,包含5个外显子和4个内含子[4],编码261个氨基酸。CD40L主要表达于活化的CD4+T淋巴细胞表面,可作用于CD40分子从而使B淋巴细胞分泌的免疫球蛋白发生类别转换(class switch recombination,CSR)。CD40L的缺陷造成免疫球蛋白CSR功能障碍,导致XHIGM患者IgG、IgA和IgE水平明显下降,而IgM水平正常或升高[5,6];Cicalese等[7]研究发现,CD40L缺乏症儿童滤泡辅助性T淋巴细胞(Tfh)和T调节细胞(Treg)细胞均降低,但滤泡调节性T淋巴细胞(Tfr)没有降低。同时CD40LG基因缺陷亦有发现少数患者存在T淋巴细胞功能障碍[6,8],因此CD40LG基因缺陷相关XHIGM患者在临床上易出现细菌及条件致病菌感染。文献报道的XHIGM患者中,各系统均可并发感染,如呼吸道感染、腹泻、口腔溃疡、脓毒症、尿路感染、脑膜炎/脑炎、皮肤组织感染及中耳炎、胆管炎等,其中呼吸道感染最为多见,中性粒细胞减少症也较为常见[2,3,6,8,9,10,11,12,13,14,15]。本报道该例患儿自婴幼儿期起反复出现呼吸道感染、曾有重症肺炎、淋巴结脓肿及中性粒细胞减少症等症状,本次以反复腹泻、胆汁淤积为主要表现,血IgM正常、IgG低下,血中性粒细胞减少,并有贫血、低蛋白血症、骨质疏松、生长落后等营养不良的表现,基因检测CD40LG基因exon5 c.506A>G(p.Y169C)突变,母亲为携带者,故可确诊为CD40LG基因突变导致的XHIGM。本例患儿表现为弥散性肝内胆管、肝总管、胆囊、胆总管扩张,文献尚未见报道。有报道XHIGM患儿可发生隐孢子虫感染引起反复腹泻后,可能上行导致硬化性胆管炎、肝硬化,甚至肝衰竭[9,12,16],本例患儿未检出隐孢子虫病原体,未针对该病原体进行治疗腹泻即好转,故隐孢子虫感染导致的胆管扩张依据不足,肝内外胆管扩张可能是XHIGM的表现之一。XHIGM伴随血嗜酸粒细胞升高未见报道,本例患儿血寄生虫抗体检查、大便寄生虫检查阴性排除寄生虫感染,骨髓穿刺未见异常,胃镜及病理排除嗜酸性胃肠炎,而血嗜酸性粒细胞明显升高,可能为XHIGM的临床表现之一,其原因及机制尚不清楚,需要进一步研究。目前已发现的CD40LG基因突变位点达100多种。包括点突变、无意突变、错义突变、插入突变、碱基缺失、剪接突变等,不同的基因突变位点可出现不同的临床表现[17]。约90%的XHIGM患者于4岁前出现临床症状[2]。本例患儿CD40LG基因突变exon5 c.506A>G为剪切突变,导致169位氨基酸有酪氨酸变为半胱氨酸,为致病性突变(rs786205606),但文献尚无该突变位点导致XHIGM的病例报道。
总之,本报道1例CD40LG基因突变所致XHIGM,患儿除具有文献报道的XHIGM常见特点外,弥散性肝内外胆管扩张、嗜酸性粒细胞明显升高是文献未报道的临床表现。c.506A>G是文献尚未见报道的CD40LG基因致病性突变。CD40LG基因突变所致的XHIGM的临床表型和基因型还需要进一步研究。
所有作者均声明不存在利益冲突

























