
溶血尿毒综合征(HUS)是儿童急慢性肾功能损害的重要病因之一,微血管性溶血性贫血、血小板减少和肾损伤是其特征性三联征。近年来,随着对HUS病因、发病机制认识的深入,HUS的分类变得更加详尽,诊断及治疗也更为精准,补体C5单抗的问世也极大地提高了HUS的救治率。现就HUS的诊断及治疗研究进展进行介绍。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
溶血尿毒综合征(hemolytic uremic syndrome,HUS)是以微血管溶血性贫血、血小板减少和肾损伤为特征表现的临床综合征,由Gasser等于1955年首次报道[1]。HUS属于血栓性微血管病(thrombotic microangiopathy,TMA)的一种,TMA是指在小血管(小动脉和毛细血管)壁形成血小板性微血栓,导致血小板消耗性减少、溶血性贫血及器官缺血和损伤的临床综合征,各种原因导致血管内皮细胞损伤是TMA发病的中心环节[2]。经典TMA包括血栓性血小板减少性紫癜(thrombotic thrombocytopenic purpura,TTP)和HUS。TTP的发病主要与含Ⅰ型血小板结合蛋白基序的解聚层白样金属蛋白酶13(ADAMTS-13)的遗传性或获得性缺陷有关,而HUS的病因、临床表现和发病机制则更为复杂。HUS好发于婴幼儿和儿童,常引起肾损害,重症HUS患儿如未能得到及时治疗,病死率高。现对HUS相关的诊断及治疗进展作一综述,以指导临床工作。
既往临床将HUS分为典型性HUS[腹泻相关性,主要指由以大肠杆菌O157∶H7为主的产志贺毒素(STX)细菌引起]和非典型性HUS(aHUS)(非腹泻相关性)[3]。随着对HUS发病机制研究的不断深入,分类也得以细化,2016儿童aHUS治疗国际共识建议将HUS分为四大类:感染诱发HUS、继发性HUS、钴胺素C缺失相关HUS以及由于先天性补体替代通路功能失调或甘油二酯酰激酶ε(DGKE)基因突变而导致的HUS[4](表1)。目前,对于aHUS的定义存在争议:大部分血液学家和肾科医师建议将aHUS一词保留给没有共存疾病或特定感染相关的HUS;而对于是否将aHUS一词限定于先天性补体和补体调节基因异常或抗补体因子H(CFH)自身抗体阳性或无基因异常相关HUS则存在争论,因为对HUS患者确认是否存在与补体相关的基因缺陷耗时耗力,可普及性不高。改善全球肾脏病预后组织(KDIGO)专家共识建议临床医师优先使用"原发性aHUS"指代强烈怀疑补体替代途径的潜在异常,并排除了继发性原因的HUS[5]。本文中的aHUS定义沿用以上最新指南共识指代存在补体成分和补体调节基因的先天性遗传异常以及DGKE突变等导致的HUS。

不同类型HUS的分类
Classification of different types of HUS
不同类型HUS的分类
Classification of different types of HUS
| 1.感染相关性HUS | ||
| 产志贺毒素大肠杆菌、肺炎链球菌、其他(流感A、H1N1、HIV) | ||
| 2.非典型HUS | ||
| (1)DKGE突变 | ||
| (2)补体替代途径异常 | ||
| ①基因突变 | ||
| ②抗CFH抗体 | ||
| (3)未发现补体或DKGE突变或抗CFH抗体的HUS | ||
| 3.继发性HUS | ||
| 骨髓移植、器官移植、恶性肿瘤、自身免疫性疾病,药物(钙调磷酸酶抑制剂,血管内皮生长因子抑制剂),恶性高血压,既往患有肾病 | ||
| 4.钴胺素C缺失相关性HUS | ||
注:HUS:溶血尿毒综合征;H1N1:血球凝集素1型神经氨酸酶1型流感病毒;HIV:人类免疫缺陷病毒;DKGE:甘油二酯酰激酶ε;CFH:补体因子H HUS:hemolytic uremic syndrome;H1N1:hemagglutinin 1 & neuraminidase 1;HIV:human immunodeficiency virus;DKGE:diacylglycerol kinase epsilon;CFH:complement factor H
在患有HUS的儿童中,产STX大肠杆菌HUS(STEC-HUS)的比例为85%~90%,肺炎链球菌HUS(SP-HUS)约为5%,aHUS为5%~10%[6]。STEC-HUS的年发病率成人约为2/10万,5岁以下儿童为6.1/10万[7]。18岁以下儿童SP-HUS的年发病率为0.06/10万[8]。新定义下的aHUS年发病率在18岁以下儿童中预估为(0.10~0.11)/100万[6]。70%的儿童aHUS在2岁前发病,约25%的儿童aHUS在6月龄之前发病[9]。而不到5%的儿童STEC-HUS发生在6月龄以下[10],因此,6月龄以下儿童发生HUS强烈提示为aHUS。婴儿钴胺素C缺乏相关HUS的发病率为(1.0~2.7)/10万[11]。
微血管内皮细胞损伤是所有类型HUS的共同特征。内皮细胞损伤的触发因素可能是外来的和暂时的,如感染、药物或肿瘤。当触发因素消除后疾病就会缓解且复发风险低。相反地内皮细胞的损伤可能是内源性或持续性的,如补体替代途径基因突变、DKGE突变或钴胺素代谢异常等。在这种情况下,HUS的复发是常见的,未经治疗的患者预后很差。
STEC-HUS的典型临床表现为3~5 d水样腹泻后发展为血性腹泻和严重腹痛,并伴恶心和呕吐,在腹泻发作2~14 d出现血小板减少症和急性肾损伤。肾外表现有神经系统症状、胰腺炎、肠坏死或穿孔、手指或脚趾坏疽、溃疡性坏死性皮肤病变、心肌梗死、缺血性心肌病[12]。STEC-HUS的发病机制为:STX与肠上皮细胞紧密结合,导致绒毛刷状缘的凋亡和破坏,随后毒素进入循环[13],在肾脏中,STX通过神经酰胺三己糖(Gb3)结合在近端小管细胞和微血管内皮细胞上,内吞作用介导毒素进入细胞内导致核糖体失活及细胞死亡[14]。此外,STEC感染后血浆中可检测到高水平的补体激活产物(Bb因子、可溶性C5b-9、补体因子),提示补体替代途径可能被激活[15]。
SP-HUS通常在肺炎球菌感染后3~13 d出现,65%~92%的儿童患有肺炎,常合并有脓胸或积液等[16]。与STEC-HUS患者相比,SP-HUS患者少尿时间长,急性透析时间长,血小板减少时间长,住院时间长,红细胞和血小板输注量多[17]。肾脏以外的并发症也有报道:胰腺炎、肢端截肢、胆囊炎、血栓形成和听力下降等[18]。
既往研究认为T抗原(Thomsen-Friedenreich抗原)暴露是SP-HUS发病过程的核心机制:SP神经氨酸酶在循环中释放并切割红细胞、血小板、肾小球内皮细胞表面的N-乙酰神经氨酸进而暴露出神秘的T抗原,这一过程称为T激活[19],T抗原暴露后与宿主预先形成的针对T抗原的IgM抗体结合进而导致SP-HUS。但近年来这一理论受到质疑:一方面抗T抗体是低滴度抗体,溶血能力弱,T活化导致红细胞溶血依据不足[20],此外,抗T抗体是一种冷反应抗体,在37 ℃时,其既不引起红细胞凝集,也不引起补体活化[21]。有报道称人半乳糖凝集素-3可能与SP-HUS发病有关[22]。据推测,在SP-HUS补体H因子结合和脱离膜的功能可能受到损害,肺炎球菌表面蛋白C(PSPC)可增强纤溶酶原的结合促进纤溶酶生成进而导致SP-HUS内皮损伤[23]。附着在PSPC上的H因子可抑制补体激活从而阻止补体介导的杀伤,从而介导了肺炎球菌的免疫逃逸[24]。目前尚不清楚补体系统失调是导致SP-HUS血栓性微血管病变的原因,还是侵袭性肺炎球菌病的结果。
aHUS发病一般是突然的,常表现为面色苍白、呕吐、乏力和嗜睡等,常伴有完全的HUS三联征,可伴水肿、少尿、高血钾和高血压,部分患者在缓解后出现急性复发表现为进行性高血压、蛋白尿和肌酐进行性升高[9]。常见的肾外表现是中枢神经系统受累(癫痫发作、视力丧失、偏瘫、头痛、意识改变、幻觉和脑病症状),心功能不全、心肌梗死、肺出血、胰腺炎和消化道出血也有报道[25]。上呼吸道感染或胃肠炎等可触发aHUS[26],因此感染后发病并不能排除aHUS的诊断,临床上有时很难确定偶然触发事件诱发的aHUS,但当患者存在暴发性发病,疾病复发或家族性发病时应引起重视。
aHUS的发病是补体蛋白基因突变或存在补体蛋白抗体的患者,经触发事件(感染、妊娠、药物治疗、恶性肿瘤、器官移植或全身疾病)引起补体替代途径不可抑制的持续激活,从而导致膜攻击复合物形成,进而导致肾脏内皮损伤、凝血级联活化和肾小球动脉微血栓形成。导致补体替代途径失调的因素主要有补体旁路调节基因(H因子、I因子、膜辅因子蛋白CD46)突变;效应基因(B因子、C3、血栓调节蛋白)突变;CFH的自身抗体形成等,但目前尚有约30%的患者没有找到已知相关基因的致病突变[27]。DKGE基因的复合或纯合突变可导致DKGE蛋白功能丧失进而导致蛋白激酶C活化,最终导致血栓前因子上调和血管内皮生长因子的下调,从而诱导血栓形成[28]。
钴胺素C缺陷病是先天性钴胺素细胞内代谢缺陷的常见亚型,其临床表现差异大,出生第1年内发病的患者往往具有更严重的表型,如进食困难、发育落后、嗜睡和肌张力低下,迟发性发病者症状通常较轻因而容易被误诊[29]。钴胺素C缺陷病肾损害常表现为血管内溶血、血尿和蛋白尿[11],也可出现肾小管间质肾炎和近端肾小管酸中毒[30]。作为一种常染色体隐性遗传病,钴胺素C缺陷病与甲基丙二酸尿症和高胱氨酸尿病C型(MMACHC)蛋白(具有脱氰酶、脱烷基酶和黄素还原酶活性)功能缺失有关,酶活性丧失导致血同型半胱氨酸和尿甲基丙二酸异常升高,异常升高的血同型半胱氨酸会损伤内皮细胞[31]。也有病例报道在儿童钴胺素C病相关HUS存在补体功能失调[32],补体缺陷可以与钴胺素C缺陷并存,具有双重机制[11]。
当临床出现血小板减少症、微血管性溶血性贫血伴或不伴器官损伤时需警惕TMA,确诊TMA后有条件地区可测定ADAMTS-13的活性以排除TTP。由于TTP在儿童中的发病率远低于成人,KDIGO指南建议儿童在测定ADAMTS-13活性的同时不应该延迟使用依库珠单抗治疗[5],但是应该小心地监测治疗无反应的征象。
在怀疑aHUS的患者中均应该常规检测STX,约5%的STEC-HUS患者无前驱腹泻,而30%补体介导的aHUS患者并发腹泻或胃肠炎。aHUS是一种危及生命的疾病,50%的aHUS患者进展为终末期肾病(ESKD),急性期死亡率显著,需要立即诊断和治疗[6]。aHUS的诊断常是在排除STEC感染和ADAMTS-13缺乏症的基础上作出的。迄今为止,还没有针对aHUS的直接诊断手段,因为正常的补体浓度或基因检测阴性均不能排除aHUS。HUS的诊断流程见图1。
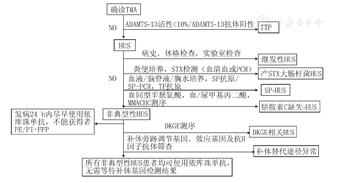
注:TMA:血栓性微血管病;ADAMTS-13:含Ⅰ型血小板结合蛋白基序的解聚蛋白样金属蛋白酶13;TTP:血栓性血小板减少性紫癜;HUS:溶血尿毒综合征;STX:志贺毒素;SP:肺炎链球菌;MMACHC:甲基丙二酸尿症和高胱氨酸尿症C型;DKGE:甘油二酯酰激酶ε;PE:血浆置换;PI:血浆输注;FFP:新鲜冷冻血浆 TMA:thrombotic microangiopathy;ADAMTS-13:a disintegrin-like metallopeptidase with thrombospondin type 1 motif 13;TTP:thrombotic thrombocytopenic purpura;HUS:hemolytic uremic syndrome;STX:Shiga toxin;SP:Streptococcus pneumoniae;MMACHC:Methylmalonic aciduria and homocystinuria type C protein ;DKGE:diacylglycerol kinase epsilon;PE:plasma exchange;PI:plasma infusion;FFP:fresh freezing plasma
支持治疗(液体和电解质管理)是所有类型HUS治疗的基石,对降低疾病死亡率有很高的临床价值。
对STEC-HUS而言支持治疗尤为重要,抗生素应用存在争议,国外学者建议谨慎使用抗生素,因为有增加STX基因表达导致STX大量释放的风险,只推荐阿奇霉素可用于菌血症儿童[35,36],而在国内普遍认为早期可使用敏感的、并避免肾毒性的抗生素[37]。美国血浆透析协会(American Society for Apheresis,ASFA)推荐,当STEC-HUS累及神经系统时可使用血浆置换术(Ⅲ级推荐),而无神经系统表现时谨慎使用(Ⅳ级推荐)[38]。对于存在危及生命并发症的患者可考虑短期免疫吸附疗法和依库珠单抗治疗[5]。依库珠单抗是结合补体C5蛋白的单克隆抗体,可抑制C5激活为C5a和C5b,并抑制膜攻击复合物的产生。
支持治疗的同时迅速开始使用抗生素(阿莫西林或三代头孢)[39],在凝集素阳性患者,应避免使用血浆和未洗脱的红细胞或血小板,因血浆中含抗T抗原抗体[6]。由于补体激活的证据,依库珠单抗已被用于治疗合并严重并发症的SP-HUS病例[40,41]。
对aHUS而言除了常规的对症支持治疗外,一旦怀疑或确诊,应在24~48 h内立即使用依库珠单抗单抗,不能获得依库珠单抗的患者建议采用血浆置换治疗[4]。CFH抗体阳性患者,血浆置换联合免疫抑制剂或糖皮质激素的疗效优于单用血浆置换,依库珠单抗也可考虑用于多器官受累的aHUS患者[42]。目前关于依库珠单抗疗程没有专家共识和指南可参考,H因子基因突变及H因子抗体阳性患者复发率相对高,没有证据证明需终身治疗,理想化的治疗目前是以最短的治疗期获得最佳的肾脏恢复而没有早期复发[43]。
对于钴胺素C缺失相关性HUS主要是肠外补充钴胺素,甜菜碱和亚叶酸可能有效[11]。器官移植或化疗药物相关的TMA应及早停用相关药物,往往预后较差。在骨髓移植相关性TMA病例中应用血浆置换治疗通常治疗效果不满意[44]。糖皮质激素及免疫抑制剂对于部分由于免疫机制异常所导致的病例可能有一定疗效[37]。
综上,HUS是因为各种内在因素和外在因素导致的补体系统功能失调、内皮细胞损伤和微血栓形成的微血管病。由于其严重的并发症和极高的死亡率,早期识别和诊断及精准治疗尤为重要。目前诊治的主要难点是,aHUS只能采取排他性诊断,基因检测过程费时费力,依库珠单抗费用昂贵,限制了其使用。但就整体而言,对补体替代途径在HUS患者发病分子机制的认识以及补体通路抑制剂药物的问世极大地提高了全球范围内HUS的救治率。
所有作者均声明不存在利益冲突

























