
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿,女,2岁11个月,因"反复发热并肝功能异常1年"就诊于江西省儿童医院消化科。此次入院前1年因"发热、呕吐3 d,精神反应差,面色苍白6 h"在江西省儿童医院PICU予呼吸机辅助通气、丙种球蛋白、头孢噻肟钠抗感染、护肝等治疗,当时肝功能明显异常[丙氨酸转氨酶(ALT)7 962 U/L,天冬氨酸转移酶(AST)11 795 U/L,白蛋白(ALB)39.2 g/L,总胆红素(TBiL)68 μmol/L],血糖低,血氨高(140 μmol/L),完善血串联质谱检查提示"多种氨基酸升高,需排除高苯丙氨酸血症、希特林蛋白缺乏症",经家属同意在北京迈基诺医学检验所完善遗传代谢基因检测(非全外显子基因测序)无阳性结果,之后复查血串联质谱及尿气相色谱均无异常,肝功能等各项指标正常后出院。入院前9个月再次因"发热、呕吐1 d"入江西省儿童医院消化科,当时亦有肝功能明显异常(ALT 3 612 U/L,AST 7 182 U/L,ALB 49.1 g/L,TBiL 22.5 μmol/L),但经丙种球蛋白调节免疫、头孢他啶抗感染、护肝降酶等治疗好转后出院,病原学检查提示存在轮状病毒感染。本次入院2 d前开始无明显诱因出现发热,具体体温家属未测,未诊治,入院1 d前出现呕吐3次胃内容物,非喷射性,偶有咳嗽,无流涕、腹泻。曾在当地医院就诊,予"头孢噻肟钠"抗感染等治疗,症状无明显缓解,遂来我院就诊,无当地肝功能检查结果。至我院门诊查血气分析提示血糖(1.7 mmol/L),拟诊"急性上呼吸道感染,低血糖"收入我院消化科。患儿系第2胎第2产,足月,因"瘢痕子宫"剖宫产娩出,出生体重3 000 g,出生时无窒息抢救史,生长发育与同龄人相符。父母体健,有一同母异父姐姐,现年13岁,体健。
入院当天我院门诊血气分析提示血糖1.7 mmol/L。腹部彩超:腹部稍胀气,肠管稍扩张,内容物多,双侧腹股沟未见明显肠管回声,肝、脾、胰、双肾、腹膜后未及明显异常。入院体格检查:体温36.5 ℃,脉搏120次/min,呼吸24次/min,体重11.5 kg,血压99/52 mmHg(1 mmHg=0.133 kPa)。无特殊面容,神志清楚。皮肤无黄染,无皮疹及出血点。咽部充血,咽峡部可见滤泡。腹软,肝脾肋下未及。心肺及神经系统查体未见异常。
诊治经过:入院后初步诊断:(1)急性上呼吸道感染;(2)急性胃肠炎;(3)低血糖;(4)中度脱水。入院当天血常规:WBC 15.29×109/L,N 86.1%,Hb 106 g/L,PLT 239×109/L,C-反应蛋白3 mg/L;肝功能:ALT 5 405 U/L,AST 9 340 U/L,ALB 39.3 g/L,TBiL 24.2 μmol/L,给予头孢他啶抗感染、补液、还原型谷胱甘肽及复方甘草酸苷护肝等对症支持治疗。但患儿仍频繁呕吐,精神反应差,伴有阵发性烦躁不安。考虑存在肝性脑病、急性肝功能衰竭,于次日上午转入PICU。转入后再次出现低血糖1.7 mmol/L,血气分析提示pH 7.392,PCO2 29.4 mmHg,PO2 128 mmHg,K+ 4.4 mmol/L,Na+ 134 mmol/L,Ca2+ 1.14 mmol/L,Cl- 101 mmol/L,实际碱剩余-6.0 mmol/L,标准碱剩余-7.1 mmol/L,乳酸6.8 mmol/L,阴离子间隙15.3 mmol/L,给予静脉泵入含糖液[糖速8 mg/(kg·min)]。考虑患儿有2次类似病史,不能排除遗传代谢性疾病,经家属同意予采集患儿及其父母外周血送北京迈基诺医学检验所行全外显子基因测序。完善凝血常规提示明显异常:凝血酶原时间216.2 s,活化部分凝血酶原时间45.2 s,国际标准化比值16.86 s,纤维蛋白原1.4 g/L;血氨139 μmol/L;IgM 0.45 g/L;EB病毒衣壳抗原IgM阴性;降钙素原1.28 μg/L。给予输注血浆支持治疗,但患儿病情进展迅速,出现意识障碍、四肢肌张力增高、呼吸浅促伴节律不齐,给予呼吸机辅助通气治疗,入院后第2天复查肝功能ALT 9 645 U/L,AST 11 275 U/L,ALB 35.1 g/L,TBiL 57.7 μmol/L;咽拭子肠道病毒通用型阳性,柯萨奇病毒A10型阳性。修正诊断:(1)急性肝功能衰竭;(2)遗传代谢病(?);(3)肠道病毒感染。给予血浆置换2次,考虑合并肠道病毒感染,IgM低,给予输注丙种球蛋白免疫支持治疗。治疗后复查肝功能、凝血常规、血氨等指标好转,神志转清楚,撤呼吸机后应家属要求转至上海复旦大学附属儿科医院继续予护肝等对症治疗,全外显子基因测序检查结果提示NBAS基因有2个杂合突变(图1),分别为c.1141delA(p.I381Sfs*10)和c.3596G>A(p.C1199Y),经家系验证分析,患儿之父母c.1141delA(p.I381Sfs*10)位点均无变异,患儿之父c.3596G>A(p.C1199Y)位点无变异,患儿之母该位点杂合变异。患儿在上海复旦大学附属儿科医院完善听力、视力、骨密度等检查均无异常后出院。出院诊断:(1)NBAS基因缺陷症;(2)小儿肝功能衰竭综合征2型。出院后在当地医院定期监测肝功能、血浆氨均正常。随访1年期间曾2次因"发热"就诊于我院消化科,入院肝功能检查均有异常,积极抗感染及输注丙种球蛋白治疗好转出院。
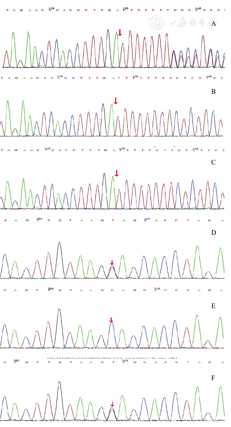
A:患儿存在c.1141delA(p.I381Sfs*10)杂合变异;B、C:患儿父母该位点无变异;D:患儿存在c.3596G>A(p.C1199Y)杂合变异;E:患儿之父该位点无变异;F:患儿之母该位点杂合变异。
NBAS基因缺陷症目前主要分为三种临床综合征:SOPH综合征、小儿肝功能衰竭综合征2型(infantile liver failure syndrome 2,ILFS2)、ILFS2与SOPH综合征重叠型[1]。此病最早于2010年发现[2],以伴有视神经萎缩的矮小症和中性粒细胞的核形态异常(Pelger-Huët)综合征为特征,是一种常染色体隐性遗传综合征,主要表现为严重的生长发育迟缓、老年样面部畸形、手脚小、智力正常、Pelger-Huët异常、视神经萎缩,统称为SOPH综合征。2015年起陆续有报道发现该基因缺陷可引起ILFS2,特征为发热期间出现急性肝功能衰竭反复发作,发作期间经治疗可完全恢复,主要临床表现为黄疸、癫痫发作、昏睡、心肌病、低血糖、高氨血症、呕吐、肝性脑病、肝脏转氨酶升高、凝血因子级联反应异常、急性肝功能衰竭[3]。本例患儿发热后肝功能明显异常,有肝性脑病、高氨血症、低血糖、凝血功能异常等表现,反复发作3次,治疗后均能恢复正常,与报道相符合。
国内NBAS基因缺陷所致疾病的报道较少,通过关键词"NBAS、小儿肝衰竭综合征2型、SOPH、ILFS2"检索中文万方数据库、中国知网、中国生物医学、维普中文科技期刊、Pubmed数据库共有中文文献报道10篇(含本文),外文文献5篇,共19例中国患儿,确诊年龄最大为17岁,最小为6个月[4,5,6,7],男女比例为6∶13,文献报道中随访无死亡病例。19例NBAS基因缺陷症中有4例以SOPH综合征为主要表现,其他15例(含本文)以反复发热后出现肝功能异常为主要表现。NBAS基因位于第2号染色体,共有52个外显子,致ILFS2基因变异分为纯合变异及复合杂合变异。目前报道的19例中国患儿均为复合杂合变异,且均来源于父母变异位点,其中常见为c.3596G>A(p.C1199Y)位点变异,共7例,本病例亦合并c.3596G>A(p.C1199Y)变异,与林建等[7]的报道一致。提示该位点可能为中国热点变异位点,与国外报道常见变异位点c.5741G>A有别。值得提出的是,本例患儿c.1141delA(p.I381Sfs*10)位点变异,目前尚无文献报道,根据美国医学遗传学与基因组学学会制定的遗传变异与分类指南初步判定为致病性变异,且该位点变异为患儿自发突变,为首次报道的新突变。
NBAS基因缺陷症诊断主要依赖于基因诊断,该患儿在第1次出现肝功能衰竭时即考虑存在遗传代谢病,但因患儿存在低血糖及血串联质谱异常,第1次完善遗传代谢基因(此基因检测主要检查以肝脏代谢及三大营养物质代谢异常疾病为主)检测无阳性结果,之后复查血串联质谱及尿气相色谱均正常,提示患儿处于危重状态时,体内各物质代谢出现紊乱,此时血串联质谱及尿气相色谱检查均可能异常,易干扰临床诊断方向。当患儿反复出现发热后肝功能异常,应警惕本病并行相关基因检测。
目前NBAS基因缺陷致ILFS2无特效治疗方法,但不同于其他原因如中毒所致肝功能衰竭患儿,虽肝功能异常明显,此类患儿经过积极护肝等治疗,均能恢复,且发作间期肝功能可正常。因其发病多由感染诱发,治疗上以积极抗感染、护肝退黄等对症治疗为主,血液净化可用于肝功能衰竭危象期病情的控制,随着年龄的增长,发作的次数及严重程度均有降低。有文献报道,此类患儿肝功能异常的严重程度与发热的热程和热峰可能有关[8]。本例患儿第1次和第3次入院热程分别为2、3 d,均因急性肝功能衰竭及肝性脑病入PICU予高级生命支持治疗,但第2次住院患儿热程为1 d,虽亦有肝功能明显异常,但无低血糖、意识改变等肝功能衰竭表现,提示及时控制体温可减少患儿发生肝功能衰竭危象的发生及改善预后[9]。本例患儿3次住院期间查IgM均低,结合相关文献报道[10],NBAS基因缺陷可表现为免疫功能异常,予丙种球蛋白输注免疫支持可有效控制感染及减少感染的发生;当患儿存在低丙种球蛋白血症时,发作间期是否需定期输注以减少感染的发生仍需进一步探讨。如经保守治疗无效,此类患儿进行肝移植为最后的选择,有文献报道1例患儿3岁时因肝功能衰竭行肝移植,随访至文献报道时间未再出现肝功能衰竭[11]。国外报道存活年龄最大为34岁[12],因此,如能及时识别此病,积极控制及预防感染,如发生肝功能衰竭,积极护肝等对症治疗,此类患儿亦有较好预后。
所有作者均声明不存在利益冲突

























