
对2019年11月南京医科大学附属儿童医院康复科诊断的1例Gillespie综合征患儿的病历资料进行回顾性分析。患儿,男,6月龄,其临床特点为运动智力落后、肌张力低下、双眼畏光、眼球震颤、双眼不能注视及追视物,眼科裂隙灯下检查提示瞳孔固定扩大、双眼虹膜部分缺损、特征性虹膜残丝,基因检测及生物信息学分析显示患儿ITPR1基因第26内含子存在1个杂合剪接突变c.3256-1G>A,为新发致病性变异,患儿父母该基因位点均未见异常。提示有双眼虹膜部分缺损、运动智力落后及肌张力低下的患儿应考虑Gillespie综合征,完善基因检测有助于早期明确诊断。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Gillespie综合征(Gillespie syndrome,GS,OMIM 206700)是一种非常罕见的常染色体单基因遗传病,1965年由Gillespie[1]首次报道并命名,其临床主要表现为双眼虹膜部分缺损、肌张力低下、非进行性共济失调和轻度至中度智力障碍。随着基因检测技术的应用,2016年发现该病的致病基因为ITPR1基因[2,3]。GS的遗传方式多数为常染色体显性遗传,少数为常染色体隐性遗传。目前该病在国内尚未见报道。现对1例GS患儿的临床及基因特点进行报道,以提高临床医师对该病的认识。
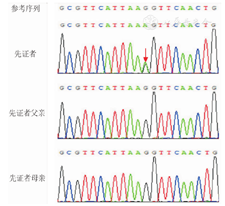
患儿,男,6月龄,因"运动智力发育落后"于2019年11月就诊于南京医科大学附属儿童医院康复科。患儿出生后数天未睁眼,在当地医院眼科检查示瞳孔固定扩大,后至南京医科大学附属儿童医院眼科就诊,诊断为虹膜部分缺损。患儿为第3胎,第2产,足月因"羊水浑浊"剖宫产,出生体质量3.3 kg,Apgar评分不详,无窒息抢救病史,母孕早期有先兆流产保胎史,余无特殊。父母非近亲结婚,否认家族遗传性神经系统疾病及眼科疾病,有一姐姐,6岁,发育正常。患儿入院时大运动落后明显,竖头不稳,不会翻身,双眼不能注视及追视物,不能主动抓物。查体:体质量8.5 kg,头围43 cm,神志清楚,头颅形态正常,前囟平软,患儿双眼畏光,双眼喜上翻(图1A),有眼球钟摆样震颤。心律齐,心音有力,未闻及明显杂音。肝肋下1 cm,脾未触及。生理反射:膝腱反射(+),原始反射:侧弯反射(+)、足趾握持反射(+),病理反射:巴氏征(+),四肢肌力正常,四肢肌张力低下,内收肌角150°,腘窝角140°,足背屈角70°,脊柱以及四肢无畸形。辅助检查:血尿粪常规、肝肾功能、电解质、心肌酶谱、血脂均未见异常。甲状腺功能正常。血串联质谱未见异常。头颅磁共振成像(MRI)平扫未见异常。骨盆正位片、普通脑电图、心电图、视听觉诱发电位均未见异常。心脏B超示卵圆孔未闭。眼科检查示双眼固视不良,角膜、前房形态正常,眼底镜检查未见异常。裂隙灯下眼前节照相可见双眼虹膜部分缺损、瞳孔固定扩大及虹膜残丝(图1B)。0~6岁儿童Gesell发育诊断量表显示,适应性、大运动2个月,精细动作、语言、个人-社交3个月。抽取患儿及患儿父母静脉血2 mL,送广州金域(医学检验)公司进行全外显子测序,对基因组DNA文库进行人类全外显子捕获,Illumina测序平台进行高通量测序,测序数据经生物信息学分析及数据库注释,并按照美国医学遗传学与基因组学会(ACMG)指南[4]对变异位点进行变异解读。结果发现患儿ITPR1基因(NM_002222.5)第26内含子存在1个杂合剪接突变c.3256-1G>A,第3256-1号核苷酸由鸟嘌呤变异为腺嘌呤。Sanger测序验证患儿父母均不携带该变异(图2)。按照ACMG指南对该变异进行分析,该变异为剪接突变,为功能缺失型变异,为致病突变(非常强致病性证据,PVS1);通过比照千人基因组数据库(1 000 Genomes)、人类基因突变数据库(HGMD)和Clinvar数据库及近10年Pubmed文献,未见c.3256-1G>A突变报道和收录(中等致病性证据,PM2);患儿父母经Sanger测序验证不携带该变异,考虑为新发突变,未经家系验证(中等致病性证据,PM6);多个蛋白功能预测软件(包括Provean、Polyphen2、Sift、Mutationtaster)预测该变异明显影响mRNA剪接(支持性致病性证据,PP3);患儿的临床表现与ITPR1基因突变导致的GS表型高度吻合(支持性致病性证据,PP4)。因此根据ACMG评级规则,c.3256-1G>A突变(PVS1+PM2+PM6+PP3+PP4)判断为未见文献报道的致病性变异。本研究征得患儿监护人知情同意,并签署知情同意书,获得医院医学伦理委员会批准(批准文号:202004034-1)。
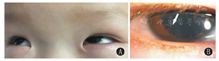
GS是一种罕见的常染色体遗传病,自1965年首次报道后,文献报道不足60例。GS病变主要累及眼睛和脑部。双侧虹膜部分缺损是识别本病最可靠的临床特征。患者均有双眼虹膜部分缺损,可见残存虹膜呈明显的扇形外观,虹膜残丝以一定的间隔延伸到前晶体表面。脑部病变主要累及小脑,婴儿早期可仅表现为肌张力低下、大运动及精细运动落后,而后逐渐出现小脑共济失调的症状,多为非进行性。GS患儿运动里程碑均显著延迟,多数在6岁后才能独走。几乎所有患者均表现出智力障碍,多数为轻度至中度。部分患者有骨骼发育异常,主要影响脊椎或手足,如脊柱侧凸、脊柱后凸、右手小指短小弯曲、第四趾骨短小、双侧扁平足等[2,3,5,6,7,8]。其他常见异常有先天性心脏病,如肺动脉或肺动脉瓣狭窄、卵圆孔未闭、房间隔缺损等[2,3,5,6]。少数患者可有消化道异常,表现为胃食管反流、肠旋转不良、剧吐[2,9]。几乎所有患者头颅MRI检查均可见不同程度的小脑萎缩,主要为小脑蚓部萎缩,少数可见脑白质异常信号。本例患儿有双眼虹膜部分缺损、特征性虹膜残丝、肌张力低下、运动智力落后、卵圆孔未闭表现,符合GS的临床特征。患儿头颅MRI未见小脑萎缩表现,共济失调症状未显现,主要是因为患儿年龄小,以后还需长期进行追踪随访。
2016年2个独立的全外显子测序项目组同时确定GS的致病基因为ITPR1基因[2,3]。ITPR1基因定位于染色体3p26.1,含63个外显子,编码1,4,5-三磷酸肌醇受体Ⅰ型(inositol 1,4,5-triphosphate receptor type 1,ITPR1)蛋白,ITPR1蛋白主要存在于滑面内质网膜上,参与组成同型四聚体钙离子通道蛋白复合物,介导钙离子从内质网释放。ITPR1基因在神经系统中尤其在小脑浦肯野细胞中高表达,在小脑的发育过程中发挥重要作用。研究证实,GS是由ITPR1基因显性杂合突变、复合杂合隐性突变或纯合隐性突变所致,通常显性杂合突变为新生突变[2,3]。目前GS患者中已报道的ITPR1基因突变类型有错义、无义、剪接、框内缺失和移码突变[2,3,5,6,7,8,9]。其中ITPR1 c.7687_7689delAGG(p.K2596del)显性杂合突变报道病例最多,为热点突变。该位点位于ITPR1基因的3′末端区域的钙转运通道的蛋白跨膜区。Gerber等[3]在体外实验发现该突变通过形成显性突变体,与野生型蛋白产生显性负效应从而抑制野生型离子通道蛋白功能,最终引起钙离子释放活性降低,提示钙离子信号通路在GS的发病过程中发挥作用。本例患儿基因检测及生物信息学分析显示ITPR1基因第26内含子存在1个杂合剪接突变c.3256-1G>A,患儿父母的该基因位点均无异常,为新发致病性变异,故符合常染色体显性遗传方式。
GS诊断主要依据患儿临床表现,如典型双眼虹膜部分缺损表型、肌张力低下、非进行性共济失调、智力障碍等,结合分子遗传学检查进一步确定诊断。需与PAX6基因变异致一种以中央凹发育不全、白内障、进行性角膜混浊和虹膜缺损为典型的广泛性眼部疾病相鉴别[10]。故所有诊断无虹膜的病例均需仔细进行临床评估,以确定有无其他眼部和眼外特征。
目前GS尚无特效治疗方法,患儿绝大多数有运动、认知落后及肌张力低下,可进行综合康复治疗,包括运动、认知功能的训练和物理理疗等,尽可能减轻发育落后的程度。需定期复查头颅MRI及应用国际合作共济失调评估量表评估患儿小脑功能。对于虹膜部分缺损,可佩带有色角膜接触镜,定期评估患儿视力。目前尚未见GS对寿命影响的报道。GS患儿父母再生育时需进行遗传咨询,因为不能排除患儿父母为生殖细胞嵌合携带者的可能。
综上所述,本研究通过报道1例GS病例,希望提高临床医师对该病的认识。临床对于有虹膜部分缺损、运动智力落后及肌张力低下的患儿,可早期完善遗传学检测,明确病因诊断,达到早期诊断和早期干预的目的,避免误诊漏诊。
所有作者均声明不存在利益冲突

























