
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
肯尼迪病(Kennedy′s disease,KD)又称X-连锁脊髓延髓肌萎缩(spinal and bulbar muscular atrophy,SBMA),由美国Kennedy等于1968年第一次报道。1991年La Spada首次发现,该病的基因缺陷在于雄激素受体基因异常扩增[1]。由于KD为罕见病,国外的发病率约为1~2/10万[2],常被误诊为肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)或其他神经肌肉疾病,国内仅有少量文献报道。陆军军医大学附属第一医院康复医学科收治1例以疑似ALS肢体功能障碍就诊康复医学科患者,经神经电生理检查发现,存在广泛慢性神经源性损害伴感觉神经动作电位波幅降低,且伴有重复神经刺激低频刺激波幅衰减和Jitter增宽等复杂电生理表现,指向KD可能,并最终通过基因检测证实。鉴于国内罕有累及神经肌肉接头KD的报道,本文总结了该病电生理特点,通过分析比较与其他易误诊疾病的电生理差异,探讨电生理检测在KD患者鉴别诊断中的价值。报道如下。
患者,男,65岁,以"四肢无力7年"就诊。7年前,患者走路时感觉双下肢无力,起病初可平路行走1 km,上楼梯需扶持;3个月后,双下肢无力加重,并出现双下肢胀痛,下蹲后起立困难,双上肢上举费力,上楼梯感腰部困痛;2年后,上述症状仍持续存在,自觉无明显变化加重,多次数家医院就诊,考虑肌萎缩侧索硬化和代谢性肌病可能,给予神经营养、改善循环和对症支持治疗后,均无效;最近1年,四肢无力进一步加重,平路行走费力,我院门诊以"肌萎缩侧索硬化"收住入神经内科,住院10 d后为行功能康复转入康复医学科。
患者自发病以来无晨轻暮重,无明显饮水呛咳、吞咽困难、构音不清,无胸闷、气短,无乳房发育及性功能减退,大小便正常,家族遗传病史(-)。
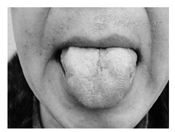
面肌有力,口周肌肉未见肌束震颤,舌肌萎缩(图1),余颅神经(-),四肢肌肉无明显萎缩,肌张力正常;颈屈肌肌力3+级,左侧三角肌肌力3级,右侧三角肌肌力3-级,双手分指肌肌力4级,双侧髂腰肌肌力3级,双侧股四头肌、胫前肌和腓肠肌肌力5级;痛觉、触觉、音叉振动觉未见异常,四肢腱反射(+),双侧Hoffmann、Babinski、Chaddock征均阴性。
血常规、风湿三项、叶酸、维生素B12水平、血沉、电解质、空腹血糖均正常;肾功能检查,尿酸测定494 μmol/L;肝功功能检查,血清丙氨酸氨基转移酶91 U/L,天门冬氨酸氨基转移酶测定84 U/L;自身抗体检测,抗SSA/Ro抗体阳性(+),余阴性;血脂,血清甘油三酯测定2.84 mmol/L;心肌酶谱,肌酸磷酸激酶为3250 U/L,肌激酶同工酶为80 U/L,乳酸脱氢酶为311 U/L;心电图检测,正常心电图;心脏彩色多普勒超声检测,主动脉硬化,左室收缩功能正常,舒张顺应性减低;四肢肌肉MRI检查,双侧下肢多发肌群异常信号,考虑肌病(图2)。左侧腓肠肌活检行肌肉病理检查,苏木精-伊红染色法(hematoxylin-eosin staining)染色,可见肌纤维大小不均匀,肌纤维坏死或分割,中心核增多,呈链状排列,肌纤维间结缔组织增加,肌间质和血管周围可见单核细胞浸润;改良Gomori染色,未见破碎红纤维(ragged red fiber,RRF);烟酰胺腺嘌呤二核苷酸四唑氧化还原酶染色,未见肌原纤维结构改变;糖原染色法(Periodic Acid-Schiff stain,PAS),未见增强。以上肌肉病理提示炎性肌病可能。
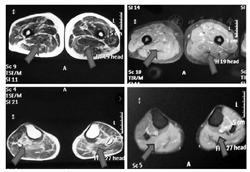
注:上图为大腿,下图为小腿,箭头所指为信号异常肌群
针极肌电图(electromyography,EMG)检测发现,双侧胫前肌、右侧腓肠肌、肱二头肌、外展小指肌、脊旁肌(T10)和舌肌均见失神经电位;左侧胫前肌、右侧肱二头肌、右侧外展小指肌、脊旁肌(T10)和舌肌运动单位动作电位(motor unit potential, MUP)时限增宽,电压增高。EMG结果提示,慢性广泛神经源性损害,详见表1。
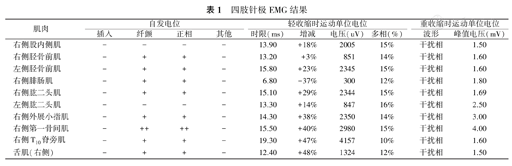
四肢针极EMG结果
四肢针极EMG结果
| 肌肉 | 自发电位 | 轻收缩时运动单位电位 | 重收缩时运动单位电位 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 插入 | 纤颤 | 正相 | 其他 | 时限(ms) | 增减 | 电压(uV) | 多相(%) | 波形 | 峰值电压(mV) | |
| 右侧股内侧肌 | - | - | - | - | 13.90 | +18% | 2005 | 15% | 干扰相 | 1.50 |
| 右侧胫骨前肌 | - | + | + | - | 13.20 | +3% | 851 | 14% | 干扰相 | 1.60 |
| 左侧胫骨前肌 | - | + | + | - | 15.80 | +23% | 2345 | 15% | 干扰相 | 1.60 |
| 右侧腓肠肌 | - | + | + | - | 6.80 | -37% | 300 | 12% | 干扰相 | 1.80 |
| 右侧肱二头肌 | - | + | + | - | 15.10 | +29% | 2344 | 15% | 干扰相 | 1.69 |
| 左侧肱二头肌 | - | - | - | - | 13.30 | +14% | 847 | 16% | 干扰相 | 2.50 |
| 右侧外展小指肌 | - | + | + | - | 14.30 | +38% | 2350 | 14% | 干扰相 | 3.00 |
| 右侧第一骨间肌 | - | ++ | ++ | - | 15.50 | +40% | 2980 | 15% | 干扰相 | 4.00 |
| 右侧T10脊旁肌 | - | + | + | - | 19.30 | +47% | 4157 | 10% | 干扰相 | 1.60 |
| 舌肌(右侧) | - | + | + | - | 12.40 | +48% | 1324 | 12% | 干扰相 | 1.50 |
神经传导检测(nerve conduction study,NCS)发现,双侧正中神经、尺神经、腓总神经、胫神经的运动神经传导速度(motor conduction velocity,MCV)在正常范围,仅双侧腓总神经刺激时引出的肌肉复合动作电位(complex muscle action potential, CMAP)波幅降低(表2);双侧正中神经、尺神经感觉神经动作电位(sensory nerve action potential, SNAP)波幅降低,双侧腓肠神经SNAP未引出。NCS结果提示,四肢感觉神经轴索受累,下肢为著(表3)。
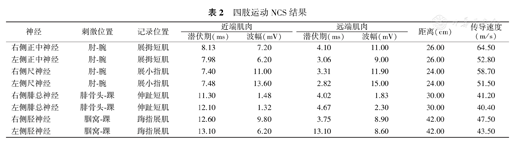
四肢运动NCS结果
四肢运动NCS结果
| 神经 | 刺激位置 | 记录位置 | 近端肌肉 | 远端肌肉 | 距离(cm) | 传导速度(m/s) | ||
|---|---|---|---|---|---|---|---|---|
| 潜伏期(ms) | 波幅(mV) | 潜伏期(ms) | 波幅(mV) | |||||
| 右侧正中神经 | 肘-腕 | 展拇短肌 | 8.13 | 7.20 | 4.10 | 11.00 | 26.00 | 64.50 |
| 左侧正中神经 | 肘-腕 | 展拇短肌 | 7.98 | 6.20 | 3.06 | 9.00 | 26.00 | 52.80 |
| 右侧尺神经 | 肘-腕 | 展小指肌 | 7.40 | 11.00 | 3.31 | 11.90 | 24.00 | 58.70 |
| 左侧尺神经 | 肘-腕 | 展小指肌 | 7.48 | 13.60 | 2.82 | 15.00 | 24.00 | 51.50 |
| 右侧腓总神经 | 腓骨头-踝 | 伸趾短肌 | 11.30 | 1.48 | 4.02 | 1.83 | 30.00 | 41.20 |
| 左侧腓总神经 | 腓骨头-踝 | 伸趾短肌 | 12.10 | 1.32 | 4.67 | 2.30 | 30.00 | 40.40 |
| 右侧胫神经 | 腘窝-踝 | 踇指展肌 | 12.60 | 9.80 | 3.75 | 8.90 | 42.00 | 47.50 |
| 左侧胫神经 | 腘窝-踝 | 踇指展肌 | 13.10 | 6.20 | 13.10 | 8.60 | 42.00 | 43.50 |
四肢感觉NCS结果
四肢感觉NCS结果
| 神经 | 刺激位置 | 记录位置 | 潜伏期(ms) | 波幅(uV) | 距离(cm) | 传导速度(m/s) |
|---|---|---|---|---|---|---|
| 右侧正中神经 | 肘 | 腕 | 2.44 | 5.90 | 14.00 | 57.40 |
| 左侧正中神经 | 肘 | 腕 | 2.42 | 10.0 | 14.00 | 57.90 |
| 右侧尺神经 | 肘 | 腕 | 2.77 | 5.10 | 14.00 | 50.50 |
| 左侧尺神经 | 肘 | 腕 | 2.42 | 7.90 | 14.00 | 57.90 |
| 右侧腓肠神经 | 踝上 | 踝 | - | - | 14.00 | - |
| 左侧腓肠神经 | 踝上 | 踝 | - | - | 14.00 | - |
注:-为未记录或未检测
单纤维肌电图(single fiber electromyography,SFEMG)、重复神经电刺激(repetitive nerve stimulation,RNS)检测发现,SFEMG——舌肌Jitter增宽(65 us)(图3);RNS——观察到双侧副神经在3 Hz、5 Hz刺激时,于斜方肌记录的CMAP波幅递减,比较第1~5个电位波幅下降比值分别为:左侧-19%、-19%;右侧:-34%、-44%;10 Hz、20 Hz刺激时,电位波幅未出现明显下降或升高,而双侧面神经、尺神经低频刺激未见波幅递减,高频刺激未见波幅明显递增。RNS和SFEMG结果提示,神经肌肉接头传递障碍。
基因检测显示,AR基因外显子1的雄激素受体基因重复数为44次,属于全突变范围,支持KD诊断。
该KD患者成年起病,主要表现为缓慢进展的肌肉无力、酸痛,由下肢发展至上肢,肌肉无力分布以近端肢带肌(骨盆带和肩胛带)为主,无深、浅感觉障碍,无共济失调,四肢腱反射(+),病理征(-)。肌酸激酶为3250 u/L,抗SSA/Ro抗体阳性,肌肉病理提示有炎性改变。
该患者有肌肉酸痛,肌肉无力以骨盆带肌和肩胛带肌为主,肌酶谱增高,肌活检有肌病改变,均提示可能为肌肉病变,如多发性肌炎、代谢性肌病等。NCS提示,有感觉神经受累,不除外多发性神经病。RNS和SFEMG提示,有神经肌肉接头传递障碍,需要鉴别重症肌无力等。该患者四肢无力7年,下肢无力起病,渐波及上肢且临床无明显感觉障碍,针极EMG呈广泛神经源损害,具有下运动神经元综合征的特征。
常见的累及下运动神经元的疾病有ALS和KD,二者均可侵犯四肢,且均为慢性病程,特别是当KD合并其它中枢神经系统病变,并出现上运动神经元体征时,更易误诊为ALS。KD是由雄激素受体(androgen receptor,AR)中雄激素受体基因重复序列扩大引起的雄激素依赖性神经肌肉遗传性疾病,下运动神经元、感觉神经和内分泌系统均可受累。虽基因检测对于KD的诊断具有重要价值[3,4],然而对于所有以下运动神经元受累为主的患者进行该基因检测其阳性率低[5]。尽管血清肌酸激酶升高亦有助于KD诊断[6],但并非特异性。而本病病理改变既可呈神经源性损害又可呈肌源性损害,给正确诊断带来极大挑战[7]。因此,在以肌无力伴肢体功能障碍就诊的康复科的患者中,相当多的KD常误诊为其他神经肌肉疾病[8],包括ALS、重症肌无力、多发性肌炎、多发性神经病、代谢性肌病等。KD呈慢性缓慢进展病程,从组织病理学上看,KD仅累及下运动神经元,而ALS则在疾病快速进展的过程中,由于细胞损伤最终会引起皮质脊髓束和脊髓侧束的损害,故上、下运动神经元均可受累。KD患者寿命正常或轻微缩短,而ALS为恶性程度高、进行性加重的运动神经元病,60%以上的患者发病3年内死亡[9],因此两者仔细甄别意义重大。
该患者在所检肌肉中,双侧胫前肌、手第一骨间肌见少量的纤颤电位和正锐波,符合KD患者病程长,以慢性损害为主。ALS病程短,进展快,侧芽生长不能充分代偿前角细胞的丢失,肌纤维得不到足够的神经再支配而处于进行性弥漫性失神经状态。因此,在静息状态下,ALS活动性失神经电位较KD明显增多。KD与ALS有着相同的病理机制,均为运动神经元损害致运动神经纤维轴索丢失,但两者在恶性程度和病情进展上大不相同。该患者所检左侧胫前肌、右侧肱二头肌及右侧外展小指肌、右T10脊旁肌和舌肌(右侧)中运动单位电位时限延长,电压增高,也符合KD轴索末梢侧芽生长相对较好,再支配也相对良好,较ALS更易见宽大MUP的电生理特点。KD可存在NCS的异常,可表现为神经传导速度减慢,远端潜伏期和F波潜伏期延长,CMAP和SNAP波幅降低,尤以感觉受累更为突出,约90%的KD患者发现SNAP波幅下降[10]。尽管KD在临床表现上感觉异常可不显著,但在神经传导和组织病理学研究中显示,大直径感觉纤维轴突有明显减少,后角或后根神经节也存在雄激素受体表达异常[6]。该患者双侧正中神经、尺神经SNAP波幅降低,双侧腓肠神经SNAP缺失符合KD以感觉神经受累突出的电生理表现,这一点有别于ALS。该患者的RNS和SFEMG均提示,有神经肌肉接头传递功能障碍。先期认为在KD中,毒性AR会损害突触前和突触后机制,导致神经肌肉突触异常,功能衰减,同时触发肌纤维膜本身过度兴奋[11,12]。Man等[13]提出了在KD中引起运动神经元死亡机制的新发现,一种可能是起源于肌肉疾病信号逆行扰乱了神经肌肉连接功能,神经肌肉接头功能障碍在组织水平上介导运动神经元的丢失和功能障碍,逆行性影响轴突转运而损害运动神经元,从而可在RNS检测中表现出CMAP异常衰减[11,12]。这也是KD患者更易误诊的又一重要原因。KD是涉及多系统多器官的基因突变性疾病,临床和其它实验室检查中均存在较大异质性,极易被误诊。电生理检测有助于KD与ALS等运动神经元病或其他神经肌肉疾病的鉴别诊断,当肌电图显示广泛神经源性损害,MUP增宽,神经传导检测发现SNAP波幅降低或缺失,这些异常结果提示为一种缓慢进展的前角细胞疾病伴有感觉神经元病变,要注意KD的可能,电生理检查的这些特征将为进一步行基因检测做出指向。
在康复医学临床,对于ALS伴肢体功能障碍患者,由于其疾病的严重性和不可逆性,且患者生存期较短,康复治疗往往侧重于现有肢体功能的维持性训练,以预防并发症为主;而对于KD后遗留功能障碍患者,由于疾病的缓慢进展,患者生存期一般影响小,利用神经电生理检测技术仔细甄别KD,并及时给予四肢功能积极的康复训练,可提高其日常生活活动能力,进而提高生活质量。KD和ALS同属运动神经元病表型,但其临床预后转归差别极大,康复目标和结局也完全不同。本研究中,KD患者的临床表现不典型,也突出了电生理检查在鉴别诊断中的必要性和意义所在。

























