
铁死亡为一种新发现的细胞程序性死亡方式,其发生机制复杂。铁过载、脂质过氧化、谷胱甘肽耗竭和丙二醛累积可通过调节细胞内长链非编码RNA、Xc-系统、GPX4、肿瘤抑制因子和核因子E2相关因子的表达诱导铁死亡。越来越多的研究表明,铁死亡与肺血管重构、急性肺损伤、慢性阻塞性肺疾病、特发性肺纤维化、肺部感染、支气管哮喘等密切相关。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
铁死亡是一种新发现的细胞死亡类型,与传统的坏死、凋亡或自噬等细胞死亡方式不同,主要由铁依赖的脂质过氧化作用引起,以线粒体收缩、线粒体膜破裂为特征[1],在细胞呼吸、能量代谢以及DNA合成和修复[2]等多种生化反应中发挥作用。肺部疾病严重威胁人类健康,越来越多的研究表明铁死亡可能与肺血管重构、肺损伤、COPD、特发性肺纤维化、肺部感染、支气管哮喘(哮喘)等相关。铁死亡通过引起炎症、内皮损伤和肺纤维化等病理过程在肺部疾病中发挥作用[3],因此,深入探索铁死亡在肺部疾病中的作用将有利于为肺部疾病的治疗提供新的思路和见解。
影响铁死亡发生机制很多,先前已证明细胞内活性氧主要作用是通过靶向多不饱和脂肪酸对生物膜造成氧化损伤,诱导铁死亡发生,而亲脂性物质(如维生素E、Ferrostatin-1和Liproxstatin-1)可抑制该过程的发生[4]。下文将从铁过载、脂质过氧化、谷胱甘肽耗竭和丙二醛累积方面阐述铁死亡发生机制(图1)。
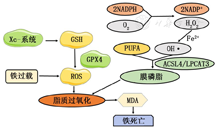
注:GSH为谷胱甘肽;GPX4为谷胱甘肽过氧化物酶4;ROS为活性氧;PUFA为多不饱和脂肪酸;OH·为羟基自由基;ACSL4为酰基辅酶A合成酶长链家族成员4;LPCAT3为溶血磷脂酰胆碱酰基转移酶3;MDA为丙二醛
铁是机体必不可少的微量元素,铁过载会对机体产生不利影响[5]。既往研究表明,铁过载可通过诱导线粒体功能障碍激活Caspase依赖的凋亡途径[6]。此后发现,铁过载导致线粒体功能障碍引起活性氧产生增加和细胞氧化应激,破坏核酸、蛋白质、脂质等大分子物质,最终引起细胞损伤或死亡[7]。
铁死亡可由铁介导[8],尽管其在铁死亡中确切机制尚不清楚,但是毫无疑问,铁代谢在铁死亡过程中发挥关键作用,原因如下:(1)铁螯合剂在体外和体内都阻止了铁死亡;(2)在铁死亡发生过程中通常细胞内不稳定铁增加;(3)铁的外源补充增加了细胞对铁死亡诱导剂的敏感性;(4)过多的血红素和非血红素铁会直接导致铁死亡;(5)几种血红素和非血红素含铁酶,如脂氧合酶、NADPH氧化酶和细胞色素负责脂质过氧化产生;(6)芬顿反应产生的铁介导活性氧增加,促进了铁死亡中脂质的过氧化作用[9]。
脂质过氧化通过两种机制发生:酶促反应和非酶促反应。该过程存在于许多细胞环境中[10],其产物是细胞中氧化应激产生的一类重要的生物分子,可直接破坏细胞膜,导致细胞功能障碍和细胞死亡[11]。有学者发现,铁死亡与脂质过氧化增加和其产物消除能力不足导致的脂质过氧化物积累有关。作为最具化学反应活性的活性氧,与Fe2+和过氧化氢通过芬顿反应引发非酶促脂质过氧化作用,引起细胞铁死亡[12]。最近研究发现,NADPH-细胞色素P450还原酶和NADPH-细胞色素B5还原酶偶联介导将电子从NADPH转移到氧气中产生过氧化氢,该过氧化氢随后通过芬顿反应与铁生成反应性羟基自由基,接着羟基自由基与膜磷脂的多不饱和脂肪酸首先经酰基辅酶A合成酶长链家族成员4或溶血磷脂酰胆碱酰基转移酶3酯化为膜磷脂(如磷脂酰乙醇胺),然后被活性氧氧化为脂质过氧化物[13],破坏膜完整性,引起铁死亡[14]。敲除酰基辅酶A合成酶长链家族成员4或溶血磷脂酰胆碱酰基转移酶3可通过消耗脂质过氧化底物来抑制铁死亡[15]。因此,推测铁死亡与脂质过氧化关系密切。
谷胱甘肽作为一种活性三肽,由谷氨酸、半胱氨酸和甘氨酸缩合而成[16]。细胞中线粒体功能障碍和(或)与铁过载有关的氧化应激增加,可导致谷胱甘肽耗竭,最终引发退行性疾病,如阿尔茨海默病、帕金森病、肌萎缩侧索硬化和缺血/再灌注损伤。
谷胱甘肽是谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)的辅助因子,即在谷胱甘肽存在时,GPX4以还原性谷胱甘肽为底物反应清除活性氧,阻止脂质过氧化反应。直到谷胱甘肽耗竭时,GPX4反应受限,亚铁离子开始分解脂质过氧化氢引发脂质过氧化,导致细胞铁死亡[17,18]。此外,抑制谷胱甘肽的抗氧化作用亦可直接抑制脂质过氧化物分解为脂质醇,诱导铁死亡发生[19]。因此,保持谷胱甘肽水平(2~10 mmol/L)对细胞保护非常重要。
考虑到细胞可能通过多种途径发生铁死亡,下面对长链非编码RNA(long non-coding RNA,lncRNA)、Xc-系统、GPX4、核因子E2相关因子(nuclear factor erythroid 2 related factor,Nrf2)和肿瘤抑制因子5种铁死亡的相关途径进行论述。
lncRNA是一类长度大于200 nt的非编码RNA,可通过与DNA、RNA或蛋白质相互作用参与表观遗传调控、转录或翻译[24],为增殖、迁移、凋亡、分化等细胞过程[25,26]中调节基因表达的关键因子。如今,越来越多的研究表明多种lncRNA可通过铁死亡调节细胞病理生理过程。据报道,在人白血病中lncRNA LINC00618表达上调可增加长春新碱诱导的铁死亡,而在肺癌中增加lncRNA LINC00336表达则可抑制铁死亡的发生[27]。此外,有文献报道在血管瘤内皮细胞中敲除lncRNA MEG8通过miR-497-5p/NOTCH2信号通路促进铁死亡发生[28]。然而与p53相关的lncRNA P53RRA可双向调节铁死亡,既可直接激活p53促进铁死亡的发生,又可抑制铁氧化或促进p53的清除阻碍铁死亡发生[29]。因此,lncRNA可通过多种途径调节铁死亡发生,有望成为诊断中的新型生物标志物。
Xc-系统为质膜胱氨酸/谷氨酸转运蛋白[30],由轻链(xCT、SLC7A11)和重链(4F2hc或CD98hc、SLC3A2)组成[31]。其中,SLC3A2作为伴侣蛋白可维持SLC7A11稳定及调节SLC7A11向质膜运输[32]。Xc-系统具有以下3种功能:(1)胱氨酸摄取以维持细胞外半胱氨酸/胱氨酸氧化还原平衡;(2)胱氨酸摄取合成谷胱甘肽;(3)谷氨酸外排[33]。研究表明,SLC7A11是调节铁过载的关键基因,抑制Xc-系统可诱导铁死亡[32,34]。使用Xc-系统的选择性抑制剂可减少细胞内半胱氨酸摄取,减少谷胱甘肽合成抑制蛋白质折叠[35]。未完全折叠蛋白质在细胞内聚集协同谷胱甘肽合成减少引起活性氧过度积累产生氧化应激,最终引起铁死亡[36,37]。
GPX4是一种重要的硒依赖型抗氧化酶[38],它可以增加细胞可塑性,为过氧化物在氧化还原信号传导过程中第二信使。如果没有硒,GPX4就会失去活性,使细胞对氧化损伤敏感度增加[39],诱导细胞凋亡和坏死的发生[40]。既往有研究表明,GPX4的主要功能是消除引起DNA损伤的过量活性氧生成[41],抑制脂质过氧化发生。此外,膜磷脂中的多不饱和脂肪酸发生脂质过氧化,直接破坏细胞膜,通过铁死亡导致细胞坏死。而GPX4还可通过减少磷脂氢过氧化表现出抗氧化活性[42],即通过氧化谷胱甘肽将脂质过氧化物转化为脂质醇,中断脂质过氧化链反应[43],以确保修复脂质过氧化引起的细胞膜破坏,从而保护细胞免于铁死亡[44]。因此,推测GPX4对铁死亡具有负调控作用,其失活会导致大量脂质过氧化并诱导铁死亡。
肿瘤抑制因子p53是一种序列特异性DNA结合蛋白[45],主要通过选择性转录调控许多靶基因来发挥其抑癌功能,从而调控细胞凋亡、细胞周期停滞、衰老、DNA修复和代谢等多种细胞过程[46]。除转录调节外,p53还与其他蛋白质直接相互作用以调节细胞凋亡和DNA修复[47]。如图2所示,p53在细胞应激状态下通过不同途径调节铁死亡[48]。一方面,p53在应激状态下过表达抑制SLC7A11以抑制胱氨酸摄取[49],使谷胱甘肽耗竭[50],最终使细胞内脂质过氧化物水平增加促进铁死亡的发生,抑制p53表达可上调SLC7A11抑制细胞铁死亡[51];另一方面,p53既可通过提高SAT1活性促进脂质过氧化促进活性氧产生,又可通过诱导谷氨酰胺酶2表达,促进谷氨酰胺分解和谷氨酸生成,从而促使铁死亡的发生[52]。此外,p53可以通过抑制二肽基肽酶4活性或诱导细胞周期蛋白依赖性激酶抑制剂1A/p21表达来抑制铁死亡[11]。
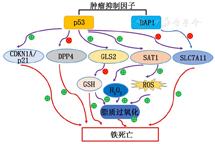
注:p53为肿瘤抑制因子p53;BAP1为BRCA1相关蛋白1;CDKN1A/p21为细胞周期蛋白依赖性激酶抑制剂1A/p21;DPP4为二肽基肽酶4;GLS2为谷氨酰胺酶2;SAT1为亚精胺/精胺N1-乙酰基转移酶1;GSH为谷胱甘肽;ROS为活性氧
然而,p53并不是唯一调节铁死亡的肿瘤抑制因子,BRCA1相关蛋白1肿瘤抑制因子也能够通过抑制SLC7A11来促进铁死亡。癌细胞中BRCA1相关蛋白1的失活导致SLC7A11上调,发挥抗铁死亡作用[53]。
Nrf2是调节细胞氧化应激反应的重要转录因子,维持氧化/抗氧化平衡。研究表明,Nrf2通过调节稳态和脂质过氧化在铁死亡中起着重要作用[54]。应激状态下Nrf2从Kelch样ECH相关蛋白1 (Kelch-like ECH-associated protein 1,Keap1)解离后转移至细胞核,并与下游抗氧化反应原件[如GPX4、血红素加氧酶1(heme oxygenase-1,HO-1)和NAD(P)H醌氧化还原酶]结合,启动下游基因的转录,防止氧化损伤[55]。此外,Nrf2的活化亦限制铁吸收,增加铁的储存,防止氧化应激和铁死亡[56]。因此,推测Nrf2的激活可能抑制铁死亡。
炎症作为许多疾病中重要的病理过程,一直是一个挑战。这种复杂的生物学反应会表现出诸如发热、疼痛、发红和肿胀等临床症状,通常由感染和组织损伤等有害刺激引起。花生四烯酸(arachidonic acid,AA)是细胞膜脂质的主要成分,通过3种途径代谢为促炎介质前体:(1)AA通过环氧合酶途径代谢为前列腺素;(2)通过脂氧合酶途径转化为白三烯和脂蛋白;(3)通过细胞色素P450途径代谢[12]。活性氧增加和氧化应激是常见的炎症特征,而丙二醛作为氧化应激的标志物,可与蛋白质、磷脂和核酸的游离氨基反应引发结构改变,引起炎症。此外,有证据表明,GPX4激活可通过减轻活性氧水平调节核因子κ B的激活和AA的氧化来减少炎症条件下细胞损害,抑制铁死亡[57]。Ferrostatin-1是一种铁死亡特异性抑制剂,可减弱炎症因子IL-1β诱导的细胞毒性、活性氧和脂质活性氧积累以及铁死亡相关蛋白表达变化,并促进Nrf2抗氧化系统的激活[58]。因此,推测在炎症相关疾病中,铁死亡可能为靶标[57]。
急性肺损伤是指由弥漫性肺间质或肺泡水肿引起的急性缺氧性呼吸功能不全或呼吸衰竭[62],发病率和病死率较高。近年来研究发现铁死亡与急性肺损伤的发病机制有关。例如,在脂多糖诱导的急性肺损伤鼠模型中除肺细胞线粒体收缩、线粒体膜破裂外,肺组织中铁过载,丙二醛、四羟壬烯醛表达增加而谷胱甘肽和GPX4减少,加入铁死亡抑制剂Ferrostatin-1后,以上与铁死亡相关因子表达被逆转[1,63,64],此外人参醇通过Keap1-Nrf2/HO-1途径减轻脂多糖诱导的急性肺损伤中铁死亡[65];在肠道缺血/再灌注损伤诱导的小鼠急性肺损伤模型中丙二醛水平显著升高,谷胱甘肽、GPX4水平显著降低,加入铁死亡抑制剂Ferrostatin-1后通过减轻肺水肿和抑制脂质过氧化来改善肠缺血/再灌注诱导的急性肺损伤。Nrf2通过促进端粒酶逆转录酶、HO-1和SLC7A11的表达来调节铁死亡,在肠缺血/再灌注诱导的铁死亡中起保护作用[66];在油酸诱导的小鼠急性肺损伤模型中观察到铁过载、谷胱甘肽水平降低、GPX4以及丙二醛含量增加,提示铁死亡在油酸诱导的小鼠急性肺损伤中起作用,但其具体机制尚不清楚;在辐射诱导的小鼠肺损伤模型中,活性氧水平增加,GPX4表达降低,加入铁死亡抑制剂Liproxstatin-1后,放射性急性肺损伤的病理改变减轻,提示铁死亡在辐射诱导的小鼠急性肺损伤中可能发挥作用[67]。
COPD是一种以支气管缓慢不可逆阻塞为特征的慢性肺部疾病,香烟烟雾为其主要危险因素[60]。有文献指出香烟烟雾可诱导COPD中支气管上皮细胞发生铁死亡,具体表现为GPX4表达下调,活性氧、脂质过氧化物和丙二醛增加[68],加入铁死亡抑制剂Ferrostatin-1、Liproxstatin-1和去铁胺后,这一过程被逆转[69]。此外,COPD中Nrf2表达显著降低,增加Nrf2可增强GPX4水平,抑制铁死亡发生。而抑制GPX4可逆转Nrf2过表达促进铁死亡,因此,靶向Nrf2/GPX4可能会抑制铁死亡[55]。有研究指出二氢槲皮素通过激活Nrf2增加SLC7A11和GPX4表达水平,抑制香烟烟雾诱导的COPD中铁死亡[70],这可能为COPD提供治疗策略。
纤维化可促使血管功能的受损,肺部疾病的特征之一就是肺纤维化[71]。在肺纤维化的发病机制中,铁死亡参与了急性放射性肺纤维化,而活性氧积累和谷胱甘肽耗竭是主要诱因。Liproxstatin-1作为铁死亡抑制剂,通过降低活性氧,限制胶原沉积的途径减轻肺纤维化,并且该抑制剂也可以缓解辐射诱导的肺纤维化中由放射引起的GPX4降低,减轻肺纤维化[56]。此外,铁死亡诱导剂Erastin通过抑制人肺成纤维细胞中GPX4表达促进成纤维细胞向肌成纤维细胞分化,促进肺纤维化。因此,推测铁死亡可能在肺纤维化中起作用,铁死亡抑制剂Liproxstatin-1通过激活Nrf2途径下调转化生长因子β1来减轻肺纤维化[72]。
特发性肺纤维化是一种慢性且严重的肺部疾病。已知成纤维细胞向肌成纤维细胞的分化与特发性肺纤维化的发病有关,有研究表明,在成纤维细胞向肌成纤维细胞分化过程中,活性氧升高,谷胱甘肽含量降低,GPX4活性降低,而加入铁死亡抑制剂Ferrostatin-1可阻止脂质过氧化抑制铁死亡,阻止向肌成纤维细胞分化[73]。此外,发现在特发性肺纤维化患者肺组织出现铁过载、脂质过氧化增加而谷胱甘肽和血红素加氧酶1增加[74]。以上提示铁死亡可能参与特发性肺纤维化进程,但具体机制尚待更多研究。
铜绿假单胞菌和结核分枝杆菌可引起肺部感染。铜绿假单胞菌通过分泌脂肪氧化酶氧化宿主细胞膜中的花生四烯酸-磷脂酰乙醇胺,于宿主支气管上皮细胞中触发铁死亡,诱导肺部感染的发生和发展。结核分枝杆菌诱导谷胱甘肽和GPX4含量降低,游离铁、线粒体超氧化物和脂质过氧化物含量增加,提示结核分枝杆菌可引起巨噬细胞铁死亡和肺组织坏死[62],加剧肺部感染。因此,铁死亡可能通过多种途径参与肺部感染。
哮喘是一种慢性炎症性气道疾病,其特征为慢性气道炎症、支气管收缩、气道高反应性和黏液分泌过多。在屋尘螨诱导的过敏性哮喘中,活性氧产生增加,谷胱甘肽耗竭,酰基辅酶A合成酶长链家族成员4、15脂氧合酶1表达增加,GPX4、SLC7A11表达下降,提示在此过程中有铁死亡发生[75]。此外,在IL-6诱导的哮喘中,支气管上皮细胞内丙二醛和活性氧释放量增加,谷胱甘肽表达水平下降,使铁过载并降低SLC7A11和GPX4表达,Ferrostatin-1加入可部分逆转脂质过氧化的影响,亦提示可能有铁死亡发生[76]。而在哮喘气道上皮细胞中抑制15脂氧合酶1表达时通过提高谷胱甘肽含量和GPX4的表达抑制铁死亡发生[77],提示15脂氧合酶1可能作为哮喘治疗的新靶点。
铁死亡作为细胞死亡方式的一种,可能在肺血管重构、急性肺损伤、COPD、特发性肺纤维化、肺部感染、哮喘等的发生和发展中发挥作用。因此,后续需更多相关研究以探索铁死亡参与肺部疾病的具体机制和通路,为肺部疾病的治疗提供参考。
所有作者声明无利益冲突

























