
肺动脉高压是一种进行性肺血管重构疾病,涉及血管壁的所有层,目前仍然无法治愈。肺动脉平滑肌细胞、内皮细胞异常增殖和凋亡抵抗是血管重构的重要特征,越来越多的研究表明,多种信号通路参与了肺动脉高压的血管重构过程,如Ras/MAPK、JAK/STAT、BMP/Smad等,确切的调控机制尚不完全清楚。本文对促进肺血管重构的几个关键信号通路进行综述,为肺动脉高压病理机制的理解及防治提供参考。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
肺动脉高压(pulmonary hypertension,PH)是一种复杂的多因素心肺疾病,以肺血管阻力和肺动脉压力持续升高为特征,最终导致右心衰竭和死亡[1,2,3]。PH病理特征包括肺血管收缩、血管重构、血栓形成和炎症等,其中进行性肺血管重构是其关键特征,涉及血管壁的所有层[2,3,4]。事实上,肺血管壁的每一种细胞类型,包括肺动脉平滑肌细胞(pulmonary artery smooth muscle cell,PASMC)、肺动脉内皮细胞(pulmonary artery endothelial cell,PAEC)、成纤维细胞、炎症细胞等,都参与了PH的病理过程,其中PASMC、PAEC异常增殖和功能障碍是肺血管重构的核心要素[2,5,6,7],在PH的发展进程中起着极其重要的作用。尽管对PH病理生理机制及治疗已经取得了很大进展,但PH仍然是不治之症,5年、7年存活率分别为57%和49%[8,9]。目前PH治疗药物主要包括前列环素类似物、内皮素拮抗剂和磷酸二酯酶抑制剂,分别靶向前列环素、内皮素和NO信号通路,扩张肺血管,改善症状,但不能逆转或延缓疾病进程、改善患者生存[10,11,12]。因此,迫切需要直接靶向血管重构机制的新治疗策略[13,14]。PASMC、PAEC增殖与凋亡,内皮-间充质转化等信号机制非常复杂[2,3,15,16,17],驱动PH血管重构的调控机制还不完全清楚。本文就PH血管重构的几个关键信号机制进行回顾性总结,为PH病理生理机制的理解及防治研究提供参考。
癌基因Ras家族成员(H-Ras、K-Ras和N-Ras等)具有调节细胞转化、致瘤性和转移的功能[18]。MAPK是丝氨酸/苏氨酸蛋白激酶,包括细胞外信号调节激酶1/2(extracellular signal-regulated kinase 1/2,ERK 1/2)、c-Jun NH2-末端激酶(c-jun NH2-terminal kinase,JNK)和p38 MAPK,通过磷酸化下游靶点以实现多种细胞活动,包括增殖、分化、代谢、迁移、存活和凋亡(图1)[19,20]。
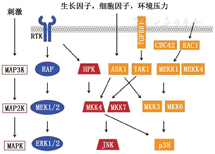
注:MAPK为丝裂原激活蛋白激酶;RTK为受体酪氨酸激酶;TGFBR1为转化生长因子β受体1;CDC42为细胞分裂周期蛋白42;RAC1为RAS相关C3肉毒杆菌毒素底物1;MAP3K为丝裂原激活蛋白3激酶;MAP2K为丝裂原激活蛋白2激酶;RAF为快速加速的纤维肉瘤;MEK1/2为丝裂原活化的细胞外信号调节激酶1/2;ERK1/2为细胞外信号调节激酶1/2;HPK为造血祖细胞激酶;MKK为丝裂原活化蛋白激酶激酶;ASK1为细胞凋亡信号调节激酶1;TAK1为转化生长因子激酶1;MEKK为丝裂原活化的细胞外信号调节激酶激酶;JNK为c-Jun NH2末端激酶
癌蛋白Ras与MAPK一起参与血小板衍生生长因子(platelet-derived growth factor,PDGF)受体、血管内皮生长因子(vascular endothelial growth factor,VEGF)受体、成纤维细胞生长因子受体信号转导通路[18,19]。Ras活化诱导MAPK激酶激酶Raf-1磷酸化,诱导MAPK激酶1/2的磷酸化,MEK 1/2通过其活化的苏氨酸和酪氨酸残基(T202/Y204)激活MAPKs ERK 1/2,丝氨酸/苏氨酸激酶ERK 1/2易位于细胞核,刺激特定的转录调节因子,诱导即早基因(c-fos、c-myc和c-jun)转录,然后活化与蛋白质和DNA合成、细胞周期进程和增殖相关路径[18]。虽然3种MAPK(ERK1/2、JNK和p38)都能促进PDGF诱导的血管平滑肌细胞增殖并在体内介导对血管损伤的反应,与其他MAPK相比,ERK对血管平滑肌细胞增殖的抑制作用最强。在缺氧暴露的PASMC中,内皮素可通过激活MAPK信号通路诱导ERK 1/2磷酸化,增强c-fos和c-jun表达,从而导致PASMC的增殖反应[20]。PDGF受体抑制剂伊马替尼可以逆转野百合碱诱导的PH和慢性缺氧诱导的PH[19]。
环境应激物、炎性细胞因子、5-羟色胺、骨形态发生蛋白受体2(bone morphogenetic protein receptor 2,BMPR2)突变体可激活p38 MAPK导致PASMC和PAEC增殖。同时,p38 MAPK活化还会触发炎症信号瀑布,在特发性PH中,炎症介质IL-6血清水平升高,抑制p38 MAPK可降低IL-6表达并逆转慢性缺氧和野百合碱诱导的PH。因此,肺血管重构可部分归因于p38 MAPK活化,即MAPK信号在增殖和炎性PH表型中起重要作用[19]。另外,磷酸化的p38 MAPK可以直接瀑布到雷帕霉素(mammalian target of rapamycin,mTOR)/核糖体p70 S6激酶(P70S6K),抑制细胞核叉头盒转录因子,促进细胞增殖,确切机制还不清楚[20]。另外,MAPK可以被促有丝分裂因子抵抗素样分子β激活促进PASMC增殖参与肺血管重构[21]。
Janus激酶(Janus kinases,JAKs)是非受体酪氨酸激酶,由4个成员组成:JAK1、JAK2、JAK3和酪氨酸激酶2[18]。在哺乳动物中,存在7种不同的信号转导和转录激活因子(signal transducers and activators of transcription,STAT)基因,它们的特征序列以及组织特异性分布能够产生特征性反应[18]。JAK/STAT通路是细胞内信号通路,可以转导各种生长因子、细胞因子和蛋白酪氨酸激酶的细胞内信号,并调节多种生理和病理过程,包括炎症、免疫反应、分化和凋亡[20]。
当配体与相应的跨膜受体结合时,受体相关的JAKs进入胞浆,导致构象改变并自动磷酸化,激活的JAKs通过其酪氨酸残基向STAT提供对接位点,STAT被磷酸化并激活。活化的STAT从受体解离,形成同源二聚体和异二聚体,并快速易位至细胞核。在细胞核与干扰素应答元件的不同启动子序列结合,调节靶基因的转录[18,20]。
缺氧和PDGF可激活PASMC中JAK/STAT信号通路,促进JAKs表达,磷酸化STAT1和STAT3,引起PASMC增殖[20]。STAT3可以整合上游信号(JAKs)并转导这些信号以维持细胞促增殖和抗凋亡表型[20]。其通过上调即早基因c-myc以及细胞周期蛋白D2、D3和A,下调细胞周期蛋白抑制因子p21和p27,促进G1/S期转变[18]。此外,PDGF激活STAT3需要活化的Src参与,而活化的STAT3还可以激活Src,显示Src/STAT3轴在调节PASMC增殖、迁移和抗凋亡中起关键作用[20]。另外,黏着斑激酶为胞浆非受体酪氨酸激酶,依赖于Src活性而被激活,在末梢肺动脉中存在Src/黏着斑激酶/STAT3通路调节PASMC增殖、抗凋亡、迁移,促进肺血管重构[20]。
磷脂酰肌醇3激酶(phosphatidylinositol 3-kinase,PI3K)在酪氨酸残基Y740或Y751位点与磷酸化的PDGF受体结合,导致磷脂酰肌醇(4,5)二磷酸磷酸化,并产生磷脂酰肌醇-(3,4,5)-三磷酸[18]。PI3K激活多个下游靶点,包括丝氨酸/苏氨酸蛋白激酶AKT、一些蛋白激酶C家族成员、P70S6K和Rho家族的小GTP酶[18]。PI3K/AKT信号通路对许多细胞功能至关重要,包括增殖、存活、分化和迁移[20]。该通路可被多种细胞因子激活,如Hif1、PDGF、磷酸酶和张力蛋白同源物(phosphatase and tensin homolog,PTEN)等[20]。AKT还能激活mTOR复合物1依赖的P70S6K 1介导细胞生长[18]。5-羟色胺激活PI3K/AKT/mTOR/P70S6K通路而诱导PASMC增殖,罗格列酮则抑制该通路而抑制PASMC增殖[18,20]。在缺氧诱导的PH模型中,PDGF激活PI3K/AKT通路,导致PASMC中cAMP反应元件结合蛋白缺失,进而诱导PASMC从收缩型向合成型转变,促进细胞增殖、迁移和去分化,导致肺血管重构[20]。此外,新的肿瘤抑制物PTEN是PI3K/AKT通路的关键下游分子,显示拮抗PI3K能力并通过抑制AKT活化而对细胞信号瀑布负反馈调节[20]。AKT 1异常激活可能在缺氧诱导的PASMC增殖过程中起重要作用,而PTEN过表达可显著抑制AKT 1磷酸化,降低缺氧诱导的PASMC增殖能力[20]。
TGF-β超家族是由多种生长因子组成,包括TGF-β配体、激活素、BMP,它们在调节细胞增殖、分化和凋亡中起重要作用[18,20,22,23]。BMP有两种类型的跨膜丝氨酸/苏氨酸激酶受体,Ⅰ型受体(BMPR1A、BMPR1B)和Ⅱ型受体(BMPR2)[23,24]。BMPR1和BMPR2与活化素受体样激酶(activin receptor-like kinase,ALK)形成异二聚体,每一个异二聚体组合都具有不同的配体亲和力和组织特异性[13,25]。BMPR2/ALK-1异源二聚体信号对微血管内皮细胞中的BMP9和BMP10具有相对特异性,而BMPR2/ALK-3/6信号对血管平滑肌细胞中BMP2或BMP4具有特异性[13]。BMPR2突变和功能丧失是PH最常见的病因[18,22,26],近75%的遗传性PH患者和25%的特发性PH患者发生BMPR2基因突变[11,13,26,27,28],而且即使在没有BMPR2突变的PH患者中BMPR2表达也会减少[11,13,14,22,28],尤其是在晚期PH患者,表明BMP信号失调是PH的共同特征[28,29],BMPR2成为PH的核心分子和分子基础[13,22]。
BMPR2与BMP配体结合而被激活,活化的BMPR2磷酸化BMPR1并与之形成异二聚体,然后激活的BMPR1磷酸化Smad1/5/8并与磷酸化的Smad4结合,形成的信号复合物易位于细胞核,调节靶基因转录[20,22,23,24,30]。在两个相似的通路机制中,Smad4是TGF-β和BMP信号通路的共有组分[20,22,24]。TGF-β和BMP通路之间的特异性在于使用了特异性的Smad亚型,BMP受体激活Smad1/5/8,而TGF-β受体激活Smad2/3[20]。BMP/Smad通路参与增殖和凋亡调节,BMPR2基因突变或表达减少,促凋亡Smad复合物形成减少,导致PASMC和PAEC促增殖和抗凋亡反应,这可能是PH病理机制的一种解释[20,23]。尽管BMP/Smad信号已被充分表征,但越来越多的证据表明MAPK在特定细胞类型中被TGF-β和BMP激活,且最终所有BMPR2突变体的共同特征是涉及p38 MAPK活化的功能获得。这些替代通路的异常活化可能对PH发病机制至关重要[22]。
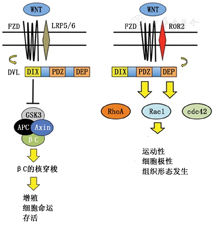
Wnt信号通路参与调控细胞增殖、凋亡、分化、存活及癌变等生理病理过程[19,20],其共同特征是Wnt配体与触发独特信号事件的膜表面受体相互作用,产生与特定Wnt通路相关的生物学结果[19]。Wnt信号通路可分为经典通路(Wnt/β-catenin,Wnt/βC)和非经典通路(Wnt/PCP通路和Wnt/Ca2+通路)[19,20]。最具特色的Wnt信号通路是Wnt/βC,靶向动态胞浆蛋白βC,通过细胞核再定位和基因表达调节来响应配体受体结合[19,20]。Wnt/βC信号通路主要由细胞外Wnt蛋白、7次跨膜卷曲蛋白受体、低密度脂蛋白受体相关蛋白5/6、散乱蛋白、糖原合成酶激酶3β(glycogen synthase kinase 3β,GSK3β)、结肠腺瘤性息肉蛋白、轴蛋白、β-catenin等信号分子组成(图2)[19]。在稳态时,轴蛋白、结肠腺瘤性息肉蛋白和GSK3β与β-catenin形成降解复合体,GSK3β对β-catenin磷酸化,使磷酸化的βC被识别并降解[19,20]。当Wnt配体与由7次跨膜卷曲蛋白受体和低密度脂蛋白受体相关蛋白5/6组成的受体复合物结合时,散乱蛋白被激活,降解复合物解体且释放β-catenin,β-catenin易位于细胞核,控制与细胞命运、增殖和存活基因相关的转录[19,20]。与Wnt/βC通路不同,Wnt/PCP通路活化与β-catenin积累无关,并且在初始激活时必须要有辅助受体酪氨酸激酶样孤儿受体2参与,散乱蛋白被激活后,激活小GTP酶Rac1、RhoA和cdc42,这些酶会引发细胞骨架重排[19]。Wnt/PCP信号负责在组织形态形成过程中调节细胞运动[19]。在Wnt/βC和Wnt/PCP之间存在串扰,这种串拢是PASMC对损伤调节的反应,而这种损伤反应可能会导致PH的功能失调并造成PASMC的行为异常[19]。此外,在PASMC和PAEC中也发现BMP和Wnt之间的信号串拢,BMP2可同时激活Wnt/βC和Wnt/PCP介导血管再生[19]。BMP2触发β-catenin活化不依赖于Wnt配体刺激,而是通过上调基因,如VEGF、存活蛋白和细胞周期蛋白D1,或诱导细胞增殖和存活[19]。BMPR2还能活化Wnt/PCP下游介质RhoA和Rac1[19]。Wnt/PCP下游Rac1和cdc42活化还可以连接MAPK通路。
Notch信号通路是高度保守信号通路,在细胞增殖、分化和凋亡中也起关键作用[20]。有4种Notch受体(Notch-1、Notch-2、Notch-3、Notch-4)和5种Delta/Serrate/Lag-2(DSL)配体,Notch-1/4主要在内皮细胞表达,而Notch-3在血管平滑肌细胞表达[20]。由于Notch受体和配体含有跨膜蛋白结构域,因此Notch通路的信号转导主要发生在相关的细胞间[20,25]。在Notch通路中,当配体结合,Notch细胞外结构域迁移并在位点2被蛋白酶ADAM10裂解,然后γ-分泌酶裂解Notch膜内锚定结构域位点3,释放各种形式的Notch细胞内结构域,然后它们全部转移至细胞核并与RBP-Jk形成活性转录复合物,调节靶基因的转录活化[20]。Notch受体与其配体间的相互作用发生在相邻细胞上,当它们在同一个细胞出现时,活化将被抑制(图3)[20]。
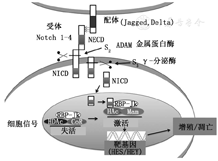
Notch通路对PH发展至关重要。其中,Notch-3几乎仅在血管平滑肌细胞中表达,参与了血管的发育和分化。在病理条件下,Notch-3调节血管平滑肌细胞收缩和合成表型之间的转换。Notch-3信号异常在血管重塑中起重要作用,血管重塑是包括PH在内的一些心血管疾病的标志。在大鼠肺组织和PH患者中,Notch-3的mRNA和Notch细胞内结构域蛋白表达增加[31]。1磷酸鞘氨醇可以通过促进YAP诱导的Notch-3表达和激活来诱导肺动脉平滑肌细胞增殖[32]。野百合碱诱导的PH大鼠模型和缺氧诱导的PH小鼠模型中,增加Notch成员蛋白(Notch-1、Notch-3、Jagged1和Herp2)表达可诱导PASMC增殖,使用腺病毒Jag1转染可以减少PASMC增殖并增强其凋亡[20]。在内皮细胞和血管平滑肌细胞BMPR2杂合子转基因小鼠中,由于内皮Notch细胞内结构域重建受阻,缺氧暴露后PH增加[33]。
近年研究发现,免疫炎症在PH中发挥重要作用,其主要通过免疫细胞在肺血管内聚集释放细胞因子和趋化因子损伤肺血管内皮细胞,引起内皮细胞凋亡增殖失衡、细胞迁移、焦亡、表型转化、PASMC增殖,最终导致血管重构[34]。炎症还可引起血管内皮细胞炎性死亡即细胞焦亡[35]。细胞焦亡作为一种程序性细胞死亡,是机体一种重要的先天性免疫反应,表现为细胞肿胀、细胞膜破裂,引起细胞内炎症因子的释放进而产生强烈的炎症反应[36]。Caspase-1是与炎症小体相关的Caspase家族的成员,裂解并促进IL-1β和IL-18的成熟,然后触发炎症反应[37]。近期有研究发现,Caspase-1、IL-1β和IL-18在PH中表达增加[36,37]。也有报道显示,在缺氧条件下,小鼠PASMC中炎症小体NLRP3激活Caspase-1[38],其通过Caspase-1/IL-18/IL-6/STAT3途径诱导PASMC增殖,从而诱导缺氧小鼠PH[36]。此外,在低氧和野百合碱诱导的PH模型中,Caspase-1抑制剂逆转了血管重构以及右心室收缩压和右心室肥大的升高[36]。以上这些发现表明,炎症反应在PH的发病机制中具有潜在的作用。
PH是一种进行性肺血管重构疾病,涉及血管壁的多种细胞、多种活性介质、信号通路和细胞因子,这些因素相互作用最终汇集成以中、小肺动脉重构为特征的病理表型,增加肺血管阻力和肺动脉压力。细胞内信号通路在血管重构过程中发挥关键的调控作用。尽管学者们已经对血管重构涉及的信号通路进行了深入研究,但精确的调控机制还有待进一步阐明。信号通路调控是一个极其复杂的调节网络,每个通路都有一个或数个反馈机制,同一通路在不同细胞的触发配体和触发顺序可能不同,不同通路间还存在相互作用,以及迄今尚未发现的信号通路或调节机制,这些都将是寻找关键的调控靶基因需要关注的方向,为PH病理生理机制的理解和临床转化提供重要依据。
所有作者声明无利益冲突

























