
X-连锁显性低血磷性佝偻病(X-linked dominant hypophosphatemic rickets/osteomalacia, XLH)是一种罕见的骨骼矿化异常性疾病,发病率为(3.9~5.0)/10万[1],是遗传性低血磷性佝偻病最常见的一型。XLH的发病机制目前认为是位于X染色体上与内肽酶同源的磷酸盐调节(phosphate-regulating endopeptidase homolog, X-linked,PHEX)基因失活性突变,引起成纤维细胞生长因子23(FGF23)清除障碍,肾脏排磷增加,尿磷增多,血磷减少,最终导致骨骼矿化障碍[2],其临床表现在儿童主要为低血磷、高尿磷、身材矮小、双下肢畸形、牙齿矿化不全等特征性佝偻病表现,而成人期则表现为骨软化症、骨关节炎、容易诱发骨折和假性骨折[3]。本文通过临床表现及遗传学分析,报道1例XLH家系,旨在提高对X-连锁显性低血磷性佝偻病的认识,以利于早期诊断,并给予合理的治疗和随访。
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
X-连锁显性低血磷性佝偻病(X-linked dominant hypophosphatemic rickets/osteomalacia, XLH)是一种罕见的骨骼矿化异常性疾病,发病率为(3.9~5.0)/10万[1],是遗传性低血磷性佝偻病最常见的一型。XLH的发病机制目前认为是位于X染色体上与内肽酶同源的磷酸盐调节(phosphate-regulating endopeptidase homolog, X-linked,PHEX)基因失活性突变,引起成纤维细胞生长因子23(FGF23)清除障碍,肾脏排磷增加,尿磷增多,血磷减少,最终导致骨骼矿化障碍[2],其临床表现在儿童主要为低血磷、高尿磷、身材矮小、双下肢畸形、牙齿矿化不全等特征性佝偻病表现,而成人期则表现为骨软化症、骨关节炎、容易诱发骨折和假性骨折[3]。本文通过临床表现及遗传学分析,报道1例XLH家系,旨在提高对X-连锁显性低血磷性佝偻病的认识,以利于早期诊断,并给予合理的治疗和随访。
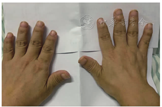
先证者为男性,33岁,30余年前因"双下肢发育畸形"就诊于外院(具体名称不详),具体诊疗过程不详,症状无改善。2020年6月因"出现双下肢疼痛"就诊于安徽医科大学第一附属医院骨科,于2020年6月4日全麻下行"左股骨截骨矫形+泰勒架固定+左胫腓骨截骨矫形外固定支架固定",因"低磷血症"转入安徽医科大学第一附属医院内分泌科就诊。病程中,患者反复出现牙齿疼痛、松动及反复牙龈发炎,曾就诊当地医院口腔科拔除牙齿(图1)。个人史:患者足月顺产,出牙、说话与同龄人相仿。自幼发现身材矮小,于16岁生长停止,最终身高143 cm。家族史:父母非近亲结婚,外公、母亲、弟弟体型均与患者相似。外公身材矮小(具体身高不详),母亲身高120 cm,弟弟身高145 cm,牙列不齐(图1)。父亲及姐姐体型、身高在正常成人范围。体格检查:身高143 cm,体重52 kg,体重指数(BMI)25.42 kg/m2。神清,精神佳,额部隆起,全口义齿,头发稀疏,无手镯征、脚镯征、肋串珠、无肋膈沟等,脊柱无畸形,双下肢"X"形。双手伸开时见两掌骨第Ⅳ掌骨远端凹陷(图2)。
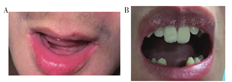
注:A:先证者口腔;B:先证者弟弟口腔
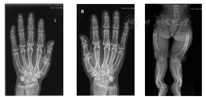
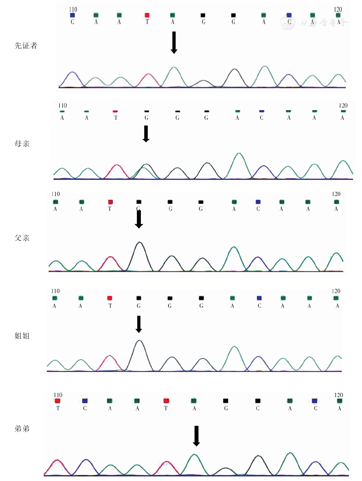
患者入院后查血磷0.43 mmol/L,低于正常范围;碱性磷酸酶126 U/L,正常范围上限;25羟维生素(25OHD)10.1 ng/ml低于正常范围;钙2.16 mmol/L,正常范围;甲状旁腺激素123 ng/L,高于正常范围;肌酐31 μmol/L、24 h尿磷13.95 mmol/L,低于正常范围;24 h尿钙5.22 mmol/24 h,正常范围(表1)。查骨密度示:腰椎L1~2低骨量,L3~4骨质疏松,股骨颈端、Wards三角低骨量,大粗隆骨质疏松,查双手及双下肢X线所见双手多发掌指骨骨质密度减低,呈膨胀性改变,部分骨端边缘骨质增生。双下肢形态扭曲,骨盆诸骨及双下肢构成骨骨密度减低,呈膨胀性改变;双股骨、右腓骨及左胫腓骨见骨质不连续,断端骨质硬化发白,右腓骨上段不规则形骨性突起,背离关节生长(图3)。基因检测结果:先证者的X染色体上内肽酶同源磷调节基因发现染色体位点22132610突变,致使核苷酸c.1208G>A,致使氨基酸p.W403X改变(图4)。
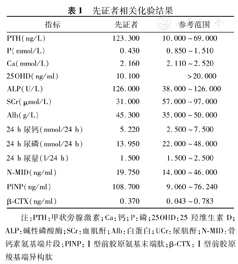
先证者相关化验结果
先证者相关化验结果
| 指标 | 先证者 | 参考范围 |
|---|---|---|
| PTH(ng/L) | 123.300 | 10.000~69.000 |
| P(mmol/L) | 0.430 | 0.850~1.510 |
| Ca(mmol/L) | 2.160 | 2.110~2.520 |
| 25OHD(ng/ml) | 10.100 | >20.000 |
| ALP(U/L) | 126.000 | 38.000~126.000 |
| SCr(μmol/L) | 31.000 | 57.000~97.000 |
| Alb(g/L) | 45.300 | 35.000~50.000 |
| 24 h尿钙(mmol/24 h) | 5.220 | 2.500~7.500 |
| 24 h尿磷(mmol/24 h) | 13.950 | 22.000~48.000 |
| 24 h尿量(l/24 h) | 1.500 | 1.500~2.500 |
| N-MID(ng/ml) | 19.750 | 14.000~46.000 |
| PINP(ng/ml) | 108.700 | 9.060~76.240 |
| β-CTX(ng/ml) | 0.370 | 0.043~0.783 |
注:PTH:甲状旁腺激素;Ca:钙;P:磷;25OHD:25羟维生素D;ALP:碱性磷酸酶;SCr:血肌酐;Alb:白蛋白;UCr:尿肌酐;N-MID:骨钙素氨基端片段;PINP:Ⅰ型前胶原氨基末端肽;β-CTX:Ⅰ型前胶原羧基端异构肽
先证者的典型临床表现:(1)身材矮小、双下肢弯曲、牙齿疾患、不同程度的骨痛。(2)实验室检查:低磷血症、碱性磷酸酶(alkaline phosphatase, ALP)升高、25OHD偏低,血钙正常,甲状旁腺激素(parathyroid hormone, PTH)正常或偏高。(3)影像学特征。(4)基因突变系列分析结果(图4)。根据以上临床表现,诊断为XLH。
先证者父亲:身高165 cm,智力正常,查体未见异常。先证者母亲:身高120 cm,脸圆,孕3产3。先证者姐姐身高160 cm,先证者弟弟身高145 cm。为进一步了解先证者家系突变基因位点的遗传方式,本研究同时对其直系亲属进行了PHEX基因位点的检测,见图4。该家系基因测序结果见表2。检测发现,患有低磷酸盐血症,X连锁显性可能性较高,患者及患者弟弟的PHEX c1208突变来自于母亲,符合X染色体疾病的遗传规律。家族遗传系谱图,见图5。
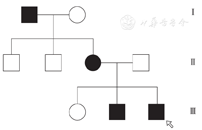
注:箭头表示先证者;□表示正常男性;■表示患病男性;○表示正常女性;●表示患病女性
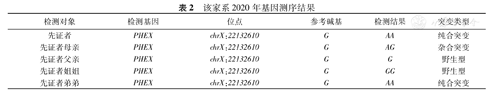
该家系2020年基因测序结果
该家系2020年基因测序结果
| 检测对象 | 检测基因 | 位点 | 参考碱基 | 检测结果 | 突变类型 |
|---|---|---|---|---|---|
| 先证者 | PHEX | chrX:22132610 | G | AA | 纯合突变 |
| 先证者母亲 | PHEX | chrX:22132610 | G | AG | 杂合突变 |
| 先证者父亲 | PHEX | chrX:22132610 | G | G | 野生型 |
| 先证者姐姐 | PHEX | chrX:22132610 | G | GG | 野生型 |
| 先证者弟弟 | PHEX | chrX:22132610 | G | AA | 纯合突变 |
先证者为33岁男性,自幼发现身材矮小,最终身高143 cm,病程中反复出现双下肢疼痛、牙齿疼痛、松动及牙龈炎症,查体:额部隆起,全口义齿,头发稀疏,双下肢"X"形。患者家族均有相似的临床特征:身材矮小(除先证者父亲、姐姐外)、"O"形腿、蹒跚步态、关节疼痛、牙列不齐。根据先证者临床表现、实验室检查、影像学特征及基因突变系列分析结果,诊断为低血磷性佝偻病。
低磷血症定义为血清磷酸盐水平低于0.8 mmol/L,低磷血症有3个主要原因[4]:由于消化吸收减少而导致体内磷的消耗,磷从细胞外腔重新分配到细胞内腔,又称为转移性低磷血症,以及由于肾脏排泄增加而导致磷的消耗(甲状旁腺功能亢进症、维生素D缺乏症、FGF23相关型肾性低磷血症等)。体内的钠磷共转运体共有3种类型,其中2型钠磷共转运体(type II sodium-phosphate cotransporter,NaPi-2)为负责体内吸收磷的主要转运体,其中NaPi-2a与NaPi-2c两个亚型主要分布于肾近曲小管,重吸收尿磷;FGF23由成骨细胞/骨细胞产生,作用于其受体,在共受体α-klotho的帮助下,抑制了近端肾小管刷状缘膜中2a型和2c型磷酸钠共转运蛋白的表达,并抑制近端肾小管磷酸盐重吸收[5]。同时FGF23通过降低1,25二羟基维生素D3[1,25(OH)2 D3]水平,抑制近端管状磷酸盐再吸收和肠道磷酸盐吸收,从而降低血清磷酸盐水平[6]。
FGF23相关型肾性低磷血症是由于体内FGF23水平过高所致,根据遗传方式可分为获得性和遗传性两大类,获得性FGF23相关型低磷血症中最常见的类型为肿瘤性骨软化症(tumor-induced osteomalacia,TIO),系肿瘤分泌过量FGF23所致。遗传性FGF23相关型低磷血症中最为常见的类型是PHEX基因突变导致的X连锁低磷血症,其发病机制目前认为是PHEX基因失活性突变,PHEX主要在成骨细胞中表达,PHEX失活后一方面增加了FGF23的表达,另一方面减少了完整的FGF23蛋白的蛋白水解裂解,导致完整的FGF23水平显著升高[7]。过量的FGF-23抑制肾脏对磷酸盐的重吸收,并且降低1,25(OH)2D水平,导致肠道磷酸盐吸收减少,均导致慢性低磷血症。常染色体隐性低磷性佝偻病(autosomal-recessive hypophosphatemic rickets,ARHR)和常染色体显性低磷性佝偻病(autosomal-dominant hypophosphatemic rickets,ADHR)是罕见的遗传性骨骼疾病,也以低磷血症为特征。ARHR与DMP1、FAM20C和ENPP1基因的失活突变有关[8]。
本文发现的PHEX基因突变c.1208G>A(p.Trp403X)为无义突变,突变位于11号外显子,导致第403位色氨酸为未知氨基酸所取代,突变点下游的阅读框漂移改变了编码产物的性质,造成PHEX功能丧失。根据家族系谱,临床考虑XLH,但不能排除ADHR。因此,采用二代测序(NGS)对先证者家族进行遗传性佝偻病基因测序,发现PHEX基因(chrX:22132610)存在突变。再用Sanger测序法对先证者及主要家族成员PHEX基因突变进行验证,显示家族中先证者、先证者弟弟、先证者母亲均存在相同基因位点突变,而未受影响的家族成员则不存在基因突变,提示此变异为致病突变。XLH为完全外显,理论上由于X染色体的随机失活,杂合子突变的女性患者病情轻,而半合子突变男性则病情较重。本文先证者家庭各成员疾病严重程度不一,相关研究提示XLH严重程度差异较大,即使在家庭成员之间也是如此,无明显的性别差异,大量PHEX失活突变可引起XLH,基因型-表型相关性不明显[9]。
XLH的生化标志是肾磷酸盐消耗引起的低磷血症、碱性磷酸酶(ALP)水平升高和完整FGF23水平升高[10]。XLH阳性家族史、ALP水平升高、与肾脏磷酸盐消耗相关的血清磷酸盐浓度降低和/或PHEX突变鉴定有助于在出生后最初几周内识别受影响的儿童。临床上,儿童期佝偻病和生长迟缓是XLH的主要特征,在成年期,患者容易患骨软化症,易发生骨折和假性骨折。患者进一步表现为退行性关节疾病、附着点障碍、肌无力、牙齿疾病(例如蛀牙和牙脓肿)以及听力损失。XLH患者的实验室结果表现为低磷血症、尿磷增加、1,25(OH)2 D3稍低或正常、血钙稍低或正常、血清碱性磷酸酶(ALP)升高和甲状旁腺激素(PTH)正常或升高。先证者实验室结果提示低磷血症、血钙正常、ALP正常范围上限、PTH异常升高。高FGF23使得1,25(OH)2 D3水平降低和血钙合成减少,但低磷血症又可刺激1,25(OH)2 D3的分泌,所以先证者血钙水平正常。血ALP水平是佝偻病活动和骨软化症的可靠生物学标志物,当佝偻病或骨软化症治疗不足时,血ALP水平升高[10]。
PHEX主要在成骨细胞中表达,对矿物结合肽/蛋白的加工有直接的基质矿化作用,XLH中PHEX的下调增加骨桥蛋白(osteopontin,OPN)沉积,这有助于局部抑制矿化。PHEX失活后一方面增加了FGF23的表达,另一方面减少了完整的FGF23蛋白的蛋白水解裂解,导致完整的FGF23水平显著升高。PHEX基因在人和小鼠之间高度保守,小鼠的PHEX在DNA水平上与人存在91%的同源性,在蛋白质水平上与人存在96%的同源性,研究表明,PHEX基因缺陷的两种同系小鼠(Hyp和Gy)已成为研究XLH和骨矿化障碍的动物模型[11]。此前,在Hyp小鼠XLH模型中,皮下注射4或16 mg/kg剂量的小鼠抗FGF23抗体可以剂量依赖的方式增加Hyp小鼠的血清磷酸盐水平及1,25(OH)2 D3水平,使肾脏磷酸盐排泄正常化,并增加NaPi-2a共转运体和1α-羟化酶在肾脏中的表达,同时降低24-羟化酶的表达[12]。Burosumab是一种针对FGF23的人单克隆抗体,抑制FGF23信号传导,从而增加肾小管磷酸重吸收。一项针对52例5~12岁XLH儿童的2期试验中证实Burosumab可改善肾小管酸盐重吸收、血清磷水平、线性生长和身体功能,缓解疼痛和佝偻病的严重程度[13]。与持续的常规治疗相比,使用Burosumab治疗的XLH患儿在佝偻病的严重程度、生长和生物化学方面显示出更大的临床改善[14]。与安慰剂相比,Burosumab显著改善XLH成人的磷酸盐代谢、骨折愈合速率及促进骨形成及骨吸收标志物的增加[15]。考虑到目前Burosumab价格及实验阶段,长期预后暂不明确,对有XLH家族史或新发的XLH患者,传统疗法仍作为一线疗法。
XLH的治疗越早效果越好,治疗的目的包括减轻骨骼畸形、增加成年身高和避免并发症发生[16]。最基本的治疗是磷酸盐合剂和1,25(OH)2 D3联合治疗,口服磷酸盐补充和活性维生素D的早期治疗可以改善佝偻病,限制牙脓肿的形成并防止进行性生长衰竭,但在很大一部分患者治疗不成功和/或伴有不良反应(如甲状旁腺功能亢进和肾钙质沉着病)。常规治疗进一步刺激FGF23水平,导致肾脏磷酸消耗,从而形成恶性循环,这可能限制其疗效[17]。这种传统的治疗方法很少能完全纠正骨骼发育,需要每日多次剂量的磷酸盐,并需要有专业知识的专家进行频繁的生化随访和仔细的剂量调整,以避免肾病钙质沉着症和继发性甲状旁腺功能亢进的潜在并发症。
综上所述,本文确定了1例家族性低血磷性佝偻病家系PHEX基因的致病突变,有助于深入了解该基因的结构与功能。本文通过基因检测确诊患者为XLH,对先证者进行及时治疗,同时也为该家系的遗传咨询和今后的产前诊断提供了可靠的依据。
所有作者均声明不存在利益冲突

























