
探讨大颗粒淋巴细胞白血病(LGL)的临床特征及实验室特点。
回顾性分析中国医学科学院北京协和医学院血液学研究所血液病医院淋巴瘤诊疗中心2004年3月至2013年5月确诊的35例LGL患者的临床及实验室特征。
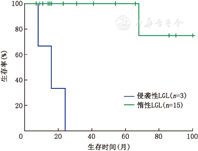
LGL起病大多隐匿,中位确诊年龄51岁,以贫血相关症状就诊者多见。11例(31.4%)患者表现为脾脏轻中度肿大,1例(2.8%)患者合并类风湿关节炎。血常规检查有贫血的患者占77.1%(27/35),外周血LGL细胞绝对值波动于(0.82~23.7)×109/L。19例(54.2%)患者表现为典型的CD3+CD57+CD56-惰性T-LGL。2例患者表现为复杂染色体核型。8例行T细胞受体可变区β链(TCRVβ)检测的患者全部表现为阳性。侵袭性LGL患者的中位总生存时间明显短于惰性LGL患者(16个月比未达到,P=0.000)。单因素分析,有B症状的患者的中位总生存时间明显低于无B症状的患者(19比45个月,P=0.039),初诊时血小板减少的患者中位总生存时间显著低于血小板正常的患者(16比42个月,P=0.000)。多因素分析时,B症状(P=0.736)和血小板减少(P=0.977)均不是LGL的独立预后因素。
我国LGL以类风湿关节炎起病者明显低于国外报道,TCRVβ检测在这一疾病诊断和鉴别诊断中具有重要意义,侵袭性LGL患者的生存期明显低于惰性患者,更需积极治疗。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
大颗粒淋巴细胞白血病(large granular lymphocytic leukemia, LGL)是一类罕见的以CD3+T及CD3-自然杀伤(NK)细胞克隆性增殖为特点的淋巴增殖性疾病。其发病机制是由于抗原的慢性刺激导致JAK/STAT3及Ras/MEK/ERK等多种信号通路组成性活化并下调凋亡及程序性死亡信号,从而促进大颗粒淋巴细胞的长期存活。典型的临床表现包括粒细胞减少、贫血以及类风湿关节炎。大部分患者最终需要治疗,但由于缺乏大规模的前瞻性临床研究,目前的治疗方案主要是基于小剂量甲氨蝶呤、环磷酰胺的免疫抑制治疗。从细胞表型来看,大颗粒淋巴细胞白血病来源于CD3+细胞毒性T细胞或CD3- NK细胞,目前世界卫生组织(WHO)分类将T-LGL列入外周T细胞肿瘤,并与侵袭性NK细胞白血病做一区分,但二者的很多分子病理机制及临床表现都类似。现就确诊的35例LGL患者的临床特点进行回顾性分析,报道如下。
收集2004年3月至2013年5月在中国医学科学院北京协和医学院血液学研究所血液病医院淋巴瘤诊疗中心确诊的35例LGL患者的临床资料,包括年龄、性别、临床表现、辅助检查,如骨髓涂片、病理、免疫组化、流式细胞术等实验室检查及患者的治疗、转归等。
所有患者均符合Sokol和Loughran[1]制定的LGL诊断标准,主要包括以下4个方面:(1)外周血中LGL细胞持续增多;(2)疾病特异性的免疫表型特点;(3)肿瘤细胞的单隆性依据;(4)疾病相关的临床表现。
采用多色流式细胞术分析患者外周血或者骨髓细胞,分析所用单克隆抗体包括抗CD2、CD3、CD4、CD5、CD7、CD8、CD16、CD56、CD57单抗。TCRVβ分析采用流式细胞术分析TCRVβ亚家族24个成员的比例。染色体核型采用R显带技术处理骨髓/外周血标本,显微镜下分析20个中期细胞。染色体核型描述依据《人类细胞遗传学国际命名体制(ISCN1995)》。
LGL的治疗指征包括:患者伴随严重感染,粒细胞缺乏,出现B症状(不能解释的低热,体温>38 ℃;盗汗;近6个月内体重减轻>10%),贫血及血小板减少。35例患者中18例接受随访的患者有治疗资料。其中5例患者确诊时无治疗指征,1例合并骨髓纤维化的患者放弃治疗,其余12例进行规律治疗,包括环孢素、泼尼松、干扰素等治疗方案。
所有数据采用SPSS 21.0统计软件进行分析。生存分析采用Kaplan-Meier方法,生存期组间比较采用Log-Rank检验。多因素预后分析采用Cox回归分析,当P<0.05时认为差异有统计学意义。
35例患者中男22例,女13例,中位确诊年龄51岁(29~79岁),年龄<60岁者24例(占68.6%)。大多数患者起病隐匿,从发病到就诊的中位时间为6.0个月(0.3~252.0个月)。多数患者因体检发现血象异常或有不适症状就诊,11例(31.4%)患者因白细胞增高就诊,13例(37.1%)患者因贫血相关症状(乏力、气促等)就诊,3例(8.5%)患者因血小板减少就诊,2例(5.7%)患者因反复感染症状就诊,1例(2.8%)患者因类风湿关节炎就诊,1例(2.8%)因下肢水肿就诊。25例(71.4%)有B症状。11例(31.4%)患者体格检查发现脾大,为轻到中度肿大,其中有1例伴有肝脏肿大。5例(14.3%)患者体格检查发现淋巴结肿大。
(1)外周血检查:35例患者外周血白细胞计数为(1.73~33.78)×109/L,血红蛋白为(36~157)g/L,血小板为(9~409)×109/L,淋巴细胞比例为0.20~0.97,32例(91.4%)患者可见大颗粒淋巴细胞。27例(77.1%)患者就诊时有中到重度贫血;23例(65.7%)患者血红蛋白<100 g/L。7例(20.0%)患者出现血小板减少。外周血LGL细胞绝对值为(0.82~23.70)×109/L。5例(14.3%)患者乳酸脱氢酶(LDH)升高。(2)骨髓细胞学检查:24例(68.6%)患者骨髓增生为活跃到明显活跃,其余患者增生减低,33例(94.3%)患者骨髓形态学可见大颗粒淋巴细胞,表现为胞质中可见中到大量细小或者粗大的嗜天青颗粒,但在2例患者中这一特点不十分明显,但经免疫表型、TCRVβ及免疫组化检查确诊为本病。骨髓淋巴细胞比例波动于0.15~0.80。(3)骨髓病理:30例(85.7%)患者骨髓病理可见淋巴细胞成灶性、散在或者弥漫性分布,免疫组化除了CD3、CD56、CD57等分子标志外可出现T细胞胞内抗原1(TIA1)、穿孔素及颗粒酶B等阳性。
35例患者行骨髓或者外周血流式细胞检查,其中19例患者具有典型的CD3+CD57+CD56-惰性T-LGL表型,3例患者具有CD3+CD56+侵袭性T-LGL表型,2例患者具有CD3-CD56+侵袭性NK细胞白血病表型,5例患者具有CD3+CD8+TCRαβ+表型,2例患者具有CD2+CD3+CD5+CD7+TCRαβ+型,CD3+CD16+CD56+CD57+ TCRγδ+表型、CD2+CD8+CD16+表型、CD3+CD4+ CD8+表型、CD2+CD3+CD5+CD7+CD8+表型各1例。在17例行染色体核型分析的患者中,有2例患者存在异常,其核型分别为45,xy,-y,-17,-18,-22[CP4]/46,XY[3]和42-46,xx,-x,-8[2]-16,-17,-19。
35例患者中8例患者行TCRVβ检测,其中6例患者发现Vβ家族某一成员比例明显增高;另外2例患者发现这些被检测的家族成员比例均降低,也间接提示尚未被检测出的家族成员比例增高。在11例行融合基因TCR重排检测的患者中,9例患者发现TCR重排阳性。
在18例有治疗资料的患者中,有5例患者确诊为惰性T-LGL后无治疗指征,至随访截止还在观察等待阶段,病情稳定。1例患者确诊T-LGL时伴骨髓纤维化,未规律治疗,8个月后因T-LGL死亡。12例患者均接受以环孢素为主的治疗方案,其中8例患者确诊后开始规律治疗,至随访截止时疾病稳定,血象恢复正常;1例患者合并纯红细胞再生障碍性贫血,至随访截止时呈输血依赖,血红蛋白维持在60 g/L;1例患者在确诊前有乳腺癌病史,规律治疗5年后因乳腺癌进展而死亡;1例侵袭性T-LGL在规律治疗2年后因疾病进展死亡;1例侵袭性NK细胞白血病患者在治疗2年后因疾病进展死亡。
所有患者随访截止时间为2013年4月30日,中位随访时间23.5个月(7.0~100.0个月)。全部患者中,失访17例,随访率为51.4%。18例患者5年的总生存率为76.7%。侵袭性T-LGL患者(3例)的中位总生存时间为16个月,惰性T-LGL患者(15例)的中位生存时间未达到,侵袭性患者的生存时间明显短于惰性患者(P=0.000,图1)。单因素分析时年龄>60岁(P=0.959)、性别(P=0.347)、血红蛋白<100 g/L(P=0.149)、LDH(P=0.607)均不影响患者的总生存。有B症状的患者的中位总生存时间明显低于无B症状的患者(19比45个月,P=0.039),初诊时血小板减少的患者中位总生存时间显著低于血小板正常的患者(16比42个月,P=0.000)。多因素分析时,B症状(P=0.736)和血小板减少(P=0.977)均不是LGL的独立预后因素。
LGL约占T/NK细胞恶性肿瘤的2%~5%。在西方国家,惰性的T-LGL大约占确诊病例的85%,男女比例相差不大,而且在老年人中多见,中位诊断年龄是60岁。NK-LGL多见于年轻患者,中位诊断年龄是39岁,在亚洲和南美洲多见。大部分T-LGL患者具有一个惰性的临床经过,中位生存时间>10年。小剂量的甲氨蝶呤、环磷酰胺及环孢素至少可以控制50%的患者的症状及血细胞减少。目前已报道有少数CD3+CD56+的T-LGL患者呈侵袭性且预后较差。NK-LGL与EB病毒感染相关,这类患者疾病进展较快并且对传统化疗反应差,中位生存时间仅为2个月[1]。惰性T-LGL患者1/3无症状,2/3患者表现为血细胞减少、脾大及类风湿关节炎[2]。这部分患者预后好,无治疗指征的可以采取"观察和等待"措施,或者免疫抑制治疗。侵袭性T-LGL临床表现为B症状、器官及淋巴结肿大及血细胞减少,这类患者应采取急性淋巴细胞白血病(ALL)样方案治疗,总体预后差。
本组病例分析显示,患者的男女比例为1.69∶1,略高于文献报道。而中位确诊年龄为51岁,低于国外报道的数据,分析可能的原因是由于人群的健康意识提高,尤其是对无治疗指征的这部分患者做出了早期确诊所致。而以贫血、白细胞增高等就诊的患者比例明显增高,并且体格检查发现脾脏肿大的患者所占比例也较高,这与文献报道基本一致。但以类风湿关节炎就诊的患者只有1例,低于国外报道。这可能与疾病的地域、人群分布有关。
35例患者中19例患者免疫表型符合典型的惰性T-LGL[3],3例患者具有侵袭性T-LGL表型,2例患者具有侵袭性NK细胞白血病表型,而其他罕见表型有CD3+CD8+TCRαβ+、CD3+CD16+CD56+ CD57+TCRγδ+、CD2+CD3+CD5+CD7+ TCRαβ+等,部分表型与近期文献报道类似[4]。有报道认为,惰性T-LGL白血病的染色体核型大多正常,不到10%的患者可表现为12p和14q的倒置,5q-以及3、8、14号三体[5]。侵袭性NK-LGL最常见的克隆性染色体异常是6q-,复杂核型也有报道[6]。本组患者中有1例惰性的T-LGL染色体核型存在异常,表现为45,xy, -y,-17, -18, -22[CP4]/46, XY[3],但这例患者经规律治疗3年后停药1年,血常规等化验完全正常。而另1例患者具有复杂核型为:42-46,xx, -x,-8[2]-16,-17,-19,由于失访无法对其进行评价。目前有关染色体核型在这一疾病诊断和预后判断中的价值的研究报道甚少,有待大规模的临床研究来证实。
由于LGL可以伴发自身免疫异常,临床上可表现为自身抗体阳性或自身免疫性疾病相关的临床症状如畏光、脱发、关节疼痛等,而在自身免疫性疾病患者中也可出现大颗粒淋巴细胞增多,二者的鉴别诊断尤为重要。因此,克隆性检测在这部分疾病的诊断中具有重要意义。传统的用于克隆性检测的主要手段是通过Southern印迹和PCR对TCR基因重排进行检测[7]。而近期研究认为TCRβ链可变区的克隆性检测可以识别75%的T细胞Vβ家族成员,目前已逐步用于临床检测[8]。本组中1例患者在就诊前有10年的类风湿关节炎病史,进一步行克隆性检测发现TCRVβ17比例明显增高。因此,这例患者应诊断为T-LGL,而不是类风湿关节炎继发的大颗粒淋巴细胞增多。NK细胞的TCR基因是不发生重排的,既往用于NK细胞的克隆性研究一般使用X-染色体失活类型(X-chromosome inactivation pattern,XCIP)作为唯一手段。近期研究认为,杀伤细胞免疫球蛋白样受体(KIR)的异常表达可以作为一个潜在的克隆性检测标志[9]。根据本结果可以看出,在判断肿瘤细胞的单克隆性时,TCRVβ检测的阳性率似乎高于传统的TCR基因重排检测的阳性率,是值得进一步推广的有助于疾病诊断的一种方法。
就总体治疗过程而言,在随访的18例患者中,大部分患者均接受以环孢素为主的治疗方案,有2例患者确诊时伴有骨髓纤维化,1例患者经过规律治疗5年后疾病稳定,而另外1例患者未行规律治疗8个月后因本病死亡。因此,对于惰性LGL患者,以环孢素为主的治疗方案疗效较好,而对伴有其他血液系统合并症的患者,应进行早期的治疗干预,争取患者的长期生存。1例侵袭性T-LGL和另外1例侵袭性NK细胞白血病的患者均在规律治疗24个月后因疾病进展死亡,而这2例患者由于年龄偏大,接受的治疗方案及强度较弱,均未给予目前推荐的ALL样方案治疗。但从总的病程来看,侵袭性LGL患者的预后明显差于惰性LGL。因此,对这部分年龄偏大、体质较弱的老年患者,如何选择一个最佳的治疗方案来获得长期生存,有待于进一步探讨。
综上所述,本组报道的35例患者的临床特点及实验室特征与国外报道基本一致,但在起病方式及人群分布亦有自己的特点。经过规范治疗后,大部分惰性LGL患者的生存期较长,而侵袭性LGL患者需要给予积极的治疗方案及治疗强度,以争取较长生存。近来,一些针对LGL的新药已逐渐进入临床试验阶段,但由于缺乏大样本的研究,其疗效有待进一步观察。

























