
探讨新生儿蛋白C缺乏症(protein C deficiency,PCD)患儿的临床特征、诊断治疗和基因特点,提高临床对PCD的认识。
对首都儿科研究所附属儿童医院新生儿科收治的1例严重新生儿PCD患儿临床资料进行回顾性分析。以“婴儿”、“新生儿”、“蛋白C缺乏”、“蛋白质C缺乏”、“暴发性紫癜”和“newborn”、“neonate”、“protein C deficiency”、“purpura fulminans”为主题词,对中国知网、万方数据库、中国生物医学文献数据库、维普生物医学数据库、Pubmed、Embase、SCI数据库收录的文献进行检索,总结已报道的新生儿PCD临床特征和基因变异特点。
本例患儿为足月女婴,生后2 d逐渐出现血小板减少、颅内出血、皮肤多发暴发性紫癜、弥散性血管内凝血、腹腔出血、高血压、门静脉及髂静脉血栓、视网膜剥离等表现,蛋白C活性<10%。基因结果回报PROC基因存在复合杂合变异,父源c.314G>T(p.C105F)和母源c.1218G>A(p.M406I),ACMG变异评级均为疑似致病变异,符合常染色体隐性遗传模式。住院6周患儿家长放弃治疗离院。检索到临床资料相对完整的新生儿病例文献共25篇,涉及患儿29例,其中男18例,女11例;早产儿4例,足月儿25例;生后7 d内起病28例;主要临床特征为皮肤暴发性紫癜及脏器血栓;记录蛋白C结果22例,范围0~25%;记录PROC基因异常结果16例,复合杂合变异10例。有预后记录的患儿22例,11例死亡,其中9例在3个月内死亡,余均遗留严重智力运动发育障碍、癫痫及失明等后遗症。
新生儿期发病的PCD主要临床表现包括暴发性紫癜、弥散性血管内凝血、全身多脏器出血或血栓等,起病急、症状重、病情进展快,预后差、病死率高,蛋白C水平检测及PROC基因检测可明确诊断。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
新生儿蛋白C缺乏症(protein C deficiency,PCD)是由于蛋白C缺乏或者活性降低导致的罕见但危及生命的急性血栓性疾病,流行病学资料估计严重PCD发病率为1/75万~1/50万,该病进展迅速,多数在新生儿期死亡[1],病死率高,预后不良,存活患儿亦多遗留严重后遗症。目前我国仅有散发个例报道,尚无用于临床治疗的蛋白C制剂,亦无肝移植治疗报道,临床对此病的认识不足。近期我院收治1例新生儿PCD,患儿存在多器官系统受累,PROC基因复合杂合变异,现报告如下,并对该病相关文献进行复习,提高临床医生对该病的认识。
1.临床资料:患儿女,生后4 d,主因“发现皮肤紫癜、血小板减少3 d,发现颅内出血2 d”入院。患儿系第2胎第2产,孕38+1周因母亲自觉无胎动并出现宫缩、羊水Ⅲ°污染剖宫产分娩,出生体重2 770 g,Apgar评分正常。母亲分娩前1天有腹泻,产后体温最高39.3℃。患儿生后第2天臀部出现紫红色硬肿瘀斑,直径约3 cm × 4 cm,血小板(platelet,PLT)29 × 109/L,C反应蛋白(C-reactive protein,CRP)10 mg/L,凝血功能提示弥散性血管内凝血(disseminated intravascular coagulation,DIC),予抗感染、人免疫球蛋白、PLT及新鲜冰冻血浆(fresh frozen plasma,FFP)输注,复查PLT升至117×109/L后复降,头颅超声示脑实质内多发高回声团,后转入我科。家族史无特殊,5岁胞兄体健。
入院查体:贫血貌,皮肤中重度黄染,前囟平,2 cm × 2 cm,张力不高。心肺腹及神经系统未见明显异常。肛周紫色硬肿瘀斑,双下肢踝关节散在红色针尖样出血点。
实验室检查:入院后血白细胞及CRP轻度升高,Hb及PLT降低;凝血酶原时间11.0~13.7 s,部分凝血活酶时间34.6~76.8 s,纤维蛋白原0.63~3.66 g/L;纤维蛋白原降解产物入院0~14 d时27.4~95.4 μg/ml,此后渐降至2.9 μg/ml;D-二聚体入院0~14 d时10.55~51.05 mg/LFEU,此后渐降至1.10 mg/LFEU。
头颅CT示双侧大脑多发实质出血,少许侧脑室出血;脑脊液WBC 260×106/L,RBC 1 780×106/L;Coomb试验±;血小板抗体阴性;ANA 1∶100,抗SSA ±,抗Ro52 ++,心磷脂抗体阴性,血管性血友病因子抗原、抗凝血酶Ⅲ正常,血管性血友病因子裂解蛋白酶(ADAMTS 13)活性稍降低。
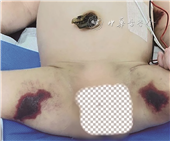
2.诊疗经过:入院后考虑患儿存在败血症及DIC,静脉应用哌拉西林他唑巴坦抗感染,FFP输注2 d停用,血小板有所上升。入院第5天患儿出现发热、贫血貌、右上臂皮肤瘀斑,血压升高,Hb及PLT降低,升级为美罗培南、万古霉素抗感染,有创辅助通气,继续纠正贫血、每天输注FFP,应用肝素及降血压治疗,病情有所改善,改无创呼吸机辅助通气。入院第10天患儿出现双侧腹股沟及右前臂瘀斑,予完善蛋白C、蛋白S检测,继续输注FFP。入院第14天患儿出现精神反应差,贫血貌、囟门张力增高,Hb、PLT下降,血不凝集,超声提示颅内出血加重,门静脉矢状部、髂静脉出现血栓。入院第16天回报蛋白C活性<10%(发色底物法,参考值60%~140%),蛋白S活性62.2%(凝固法,参考值63.5%~149%),予每天输注FFP 2次,患儿病情逐渐好转,皮肤紫癜瘀斑逐渐结痂脱落,入院30 d复查头颅MRI提示双侧大脑实质出血(亚急性期),幕上脑室扩张显著,眼底检查提示白曈、瞳孔不规则、晶体混浊、视网膜剥离,提示双眼视力丧失。入院41 d基因回报PROC基因存在一组复合杂合变异,符合遗传性PCD诊断。住院6周患儿家长放弃治疗离院。患儿生后2月余电话随访,离院后未行特殊治疗,皮肤无新出紫癜,无抽搐表现,后失访。患儿暴发性紫癜表现见图1。
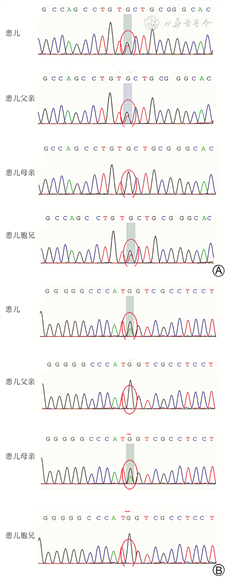
3.基因分析:取患儿及其父母、胞兄外周血进行全外显子基因组检测,结果见图2,提示PROC基因存在一组复合杂合变异,一处为c.314G>T,导致氨基酸改变p.C105F(半胱氨酸>苯丙氨酸);另一处c.1218G>A,导致氨基酸改变p.M406I(甲硫氨酸>异亮氨酸)。经Sanger测序验证,患儿父亲携带单杂合变异c.314G>T,母亲携带单杂合变异c.1218G>A,兄长携带单杂合变异c.314G>T。家系验证结果符合常染色体隐性遗传,与遗传性PCD的遗传模式吻合。
新生儿PCD报道罕见,以“婴儿”、“新生儿”、“蛋白C缺乏”、“蛋白质C缺乏”、“暴发性紫癜”和“newborn”、“neonate”、“protein C deficiency”、“purpura fulminans”为主题词,对中国知网、万方数据库、中国生物医学文献数据库、维普生物医学数据库以及Pubmed、Embase、SCI数据库收录的文献进行检索,临床资料记录详尽的报道共25篇,总计29例患儿,其中男18例,女11例;早产儿4例,足月儿25例;生后7 d内起病28例;主要临床特征为皮肤暴发性紫癜及脏器血栓,其中暴发性紫癜26例(89.7%),脏器血栓3例(10.3%),两者合并13例(44.8%);出血18例,其中颅内出血14例(48.3%),眼内出血(眼底及玻璃体内)12例(41.4%),同时存在8例(27.6%);蛋白C水平有记录共22例,范围0~25%,最低测不出,蛋白C<10% 19例,≥10% 3例;有PROC基因异常记录16例,复合杂合变异10例,纯合变异2例,4例变异类型不详。有预后记录的患儿22例,11例死亡,其中9例在3个月内死亡;余11例遗留严重的智力运动发育障碍、症状性癫痫及失明;推测7例放弃治疗,无预后记录的患儿死亡可能性大。
蛋白C是一种在肝脏合成的维生素K依赖性抗凝蛋白,与蛋白S聚集在血小板膜上水解灭活血浆中的凝血因子Va和Ⅷa,从而发挥抗凝功能。蛋白C随年龄增长而升高,约6月龄时达到成人水平[2]。早产儿蛋白C活性是成人的7%~18%[3],足月儿为20%~40%[4]。新生儿PCD是由于蛋白C缺乏或活性下降导致的血栓性疾病,临床特征为暴发性紫癜、DIC及脏器血栓或出血,部分患儿可在胎儿期出现血栓发作[5],如颅内血栓、脑积水及玻璃体内出血等表现[6]。暴发性紫癜为短时间内在微循环系统形成广泛的血凝块[7],进而出现出血性皮肤坏死、瘀点、瘀斑及周围炎症,通常在生后2~12 h内出现,臀部、大腿、小腿常受影响,手指、足趾也可受累[8],多为不同部位散发,也可同一部位好转后反复发作[9],可出现水泡破溃坏死和坏疽,甚至截肢。PCD患儿暴发性紫癜的发作可能由感染、创伤或手术引发[10],罕见动脉血栓[11]。PCD患儿脏器血栓可见于肾静脉、玻璃体静脉、髂静脉及大脑后动脉等[12],出现玻璃体出血和视网膜血栓、视网膜脱离、脑出血、脑室扩张、脑积水等严重并发症,可导致部分或完全失明甚至死亡。文献检索发现PCD患儿起病早,29例患儿出现暴发性紫癜症状26例,出血表现18例。
蛋白C由PROC基因编码,位于染色体2q13-q14,由9个外显子和8个内含子组成[13],外显子9编码FVa的催化位点和结合位点对蛋白C的功能尤为重要[14]。2019年文献已描述的PROC基因变异超过370个[15],目前HGMD数据库更新的PROC基因变异共501个,以错义变异为主,可导致蛋白C数量降低(Ⅰ型)或活性降低(Ⅱ型),大多数为Ⅰ型[16]。蛋白C缺乏分为先天性和后天性,先天性即杂合变异或者纯合变异引起,蛋白C活性水平显著降低,临床表现严重;后天性一般因蛋白C消耗增加,如B族溶血链球菌感染、DIC、急性静脉血栓形成、抗磷脂抗体阳性、心脏或其他手术等[17],或者合成减少,包括严重肝功能障碍、半乳糖血症、严重先天性心脏病及华法林或其他维生素K拮抗剂治疗等[8]。
本例患儿母亲产前存在感染,生后出现皮肤瘀斑及PLT减少、颅内脑实质多发出血,后出现皮肤暴发性紫癜、多处颅内出血、门静脉、髂静脉血栓、玻璃体出血等,蛋白C活性<10%,基因回报PROC基因存在c.314G>T和c.1218G>A杂合变异,可诊断遗传性PCD。c.314G>T(p.C105F)评级为疑似致病变异,未曾报道,相同氨基酸位置曾有c.313T>C(p.C105R)被报道为致病性变异[18],推测c.314G>T(p.C105F)可能因类似机制导致蛋白C数量降低即Ⅰ型PCD。c.1218G>A(p.M406I)已被HGMD数据库收录为致病性变异,国内外文献都有过报道[19, 20],依据ACMG指南,被评为疑似致病变异。该位点位于9号外显子,是蛋白C反应中心区,该区变异会使蛋白空间立体结构形成受阻,干扰蛋白C的稳定性和正常折叠,导致蛋白C功能失活诱发血液高凝状态[21, 22],约占日本静脉血栓栓塞症患者PROC变异的8%[23],也是导致韩国Ⅰ型PCD最常见的两个基因变异之一[21]。2017年日本关于PROC基因复合杂合变异患者的研究中有15例胎儿及新生儿,14例基因变异位于9号外显子,其中1例与本例患儿变异位点一致[24]。综上所述,本例患儿两个变异位点均可导致蛋白C数量明显减少,即Ⅰ型PCD,尤其9号外显子变异致病性可能更强,因此遗传基础决定了患儿临床表现严重,预后不良。
目前尚无治疗新生儿遗传性PCD的标准指南及研究。补充外源性蛋白C为治疗关键,FFP或人类蛋白C浓缩制剂已成功用于婴儿替代治疗[25],在美国和加拿大于2007年被食品药品管理局批准用于PCD患者,也有应用胰岛素泵持续泵入治疗的病例[26]。人类蛋白C浓缩制剂尚未在国内上市。FFP剂量为每12 h 10~20 ml/kg,频繁给予FFP可能导致高蛋白血症、高血压、静脉通路丧失以及病毒感染等。此外也有FFP联合血栓调节蛋白治疗PCD的报道[27],也有口服抗凝药物依杜沙班治疗新生儿PCD的报道[28]。肝移植可纠正纯合性蛋白C缺乏,研究报道已经在4例患儿中使用,3例存活[29, 30, 31]。
新生儿期发病的PCD临床可见暴发性紫癜、DIC、多发性血栓及脏器出血表现。本病起病急,临床症状重,蛋白C水平或活性检测及PROC基因检测可明确诊断,早期予FFP输注可改善凝血功能,需终生治疗,预后欠佳,病死率高。
所有作者均声明不存在利益冲突

























