
CIC重排肉瘤是一种具有高度侵袭性的小圆细胞肉瘤,除了常见的DUX4外,还可能存在FOXO4、LEUTX、NUTM1和NUTM2A等极少数融合伴侣。肿瘤多发生于深部软组织,极少见于中枢神经系统。2021年8月深圳市人民医院神经外科收治1例颅内原发CIC-NUTM1重排肉瘤成人女性患者,其病变位于右侧脑室,经手术全切除后患者恢复良好。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
CIC重排肉瘤是一种具有高度侵袭性的小圆细胞肉瘤,组织学上类似于尤文肉瘤,但缺少EWSR1基因易位,遗传上显示特征性的CIC基因(19q13)重排[1]。肿瘤多发生于深部软组织,其次为内脏,极少发生于中枢神经系统[2]。2021年8月深圳市人民医院神经外科收治1例成年女性原发颅内CIC-NUTM1重排肉瘤患者,经手术治疗效果良好。现结合文献,探讨该病的临床表现、诊断和治疗,以期为其临床诊治提供参考。
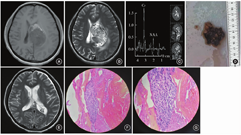
患者 女,45岁,因"右侧肢体麻木、无力半个月余,头晕2 d"入院。入院体格检查:神志清楚,对答切题,遵嘱动作,格拉斯哥昏迷评分(GCS)为15分。双侧瞳孔等圆、等大,直径约为2.5 mm,对光反射灵敏。颈强直(-),四肢肌力、肌张力检查未见明显异常。生理反射存在,病理反射未引出。半个月前于外院门诊行头颅CT检查显示左侧侧脑室内一占位性病变,最大直径约为3.5 cm。入院后第5天,患者的头晕症状明显加重,并出现嗜睡,予20%甘露醇静脉滴注后症状缓解。急诊行头颅MRI、弥散加权成像(DWI)和磁共振波谱(MRS)检查,MRI结果提示:左侧侧脑室体部可见一类圆形异常信号影,边界尚清,大小约为60 mm×43 mm×40 mm,病灶T1加权成像(T1WI)呈稍低信号,前缘可见斑片状高信号灶,T2加权成像(T2WI)呈混杂信号,以稍高信号为主,其内可见多发小囊变区(图1A,B)。DWI可见明显弥散受限,病灶周围可见大片水肿带,中线结向右侧偏移约12 mm。MRS结果显示,病灶区域乙酰天冬氨酸(NAA)峰明显降低,胆碱(Cho)峰明显升高,NAA/(Cho+肌酐)比值倒置(图1C)。考虑到肿瘤生长迅速,于入院后第2天行肿瘤切除术。在气管插管全身麻醉下,取右侧顶部"马蹄"形切口,经顶下小叶皮质造瘘进入4 cm后,可见血运丰富的鱼肉样肿瘤组织,质地不硬,伴有瘤内出血(图1D)。予快速瘤内减压,以吸引器和取瘤镊沿肿瘤边缘探查,发现肿瘤供血血管后予以电凝,打开侧脑室壁,彻底清除脑室内残留的血肿。术后第2天复查MRI显示肿瘤完整切除(图1E)。术后HE染色结果显示,病变由异型细胞构成,可见多核瘤巨细胞和核分裂象,以及大片坏死及横纹肌样细胞,间质可见黏液样变性(图1F,G)。术后免疫组织化学结果提示:INI-1(未缺失)、CD99(-)、Syn(-)、CD34(-)、STAT6(-)、BRAFV6OOE(-)、SSTR2(少灶+)、MyoDl(-)、Myogenin(-),病变符合(颅内)高级别肿瘤。鉴于根据免疫组织化学结果未能明确其病理学分型,行高通量基因检测(NGS二代测序)明确有无特殊的分子遗传学特征,分子检测结果提示:体细胞变异CIC-NUTM1融合(+),病变符合(颅内)CIC重排肉瘤。术后患者未诉特殊不适,无抽搐发作,左侧肢体肌力为Ⅳ+级,余无明显神经系统功能障碍。术后于外院继续行放射治疗。
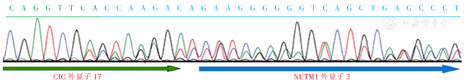
CIC重排肉瘤中,除了常见的DUX4(占所有患者的95%)外,还有FOXO4、LEUTX、NUTM1和NUTM2A等融合伴侣,约见于5%的患者中[3]。这些肿瘤的组织学形态和免疫表型与CIC-DUX4融合肉瘤基本一致,CIC-NUTM1融合肉瘤更多发生于儿童的中枢神经系统[4]。Le Loarer等[4]共报道了6例CIC-NUTM1融合患者,其中3例为发生于中枢神经系统的儿童和青少年,仅1例为发生于侧脑室三角区的22岁女性患者。另外,还有少量病例报道,CIC-NUTM1重排肉瘤可涉及脑外组织或器官,包括骨骼、软组织和内脏[5,6]。此外,还有作者发现,以CIC-NUTM1融合为特征的肉瘤常发生于中轴骨骼[7]。本例患者的分子基因检测结果即显示为体细胞变异的CIC-NUTM1融合,其为45岁女性,肿瘤发生于侧脑室体部,类似病例在以往文献中尚未见报道。
CIC重排肉瘤是最近新定义的脑肿瘤类型,属于发生于脑膜的非脑膜上皮源性间叶肿瘤,目前尚属未确定分化方向的恶性肿瘤[8]。目前对于CIC-NUTM1肉瘤的精确分类尚有争议,Mangray等[6]认为,将CIC-NUTM1肉瘤归类为NUT癌或许比归类为CIC重排肉瘤更为准确。目前的常规免疫组织化学检测结果仅能确定其是否为高级别肿瘤,不能明确病理学分型,需行高通量基因检测(MGS二代测序)明确有无特殊的分子遗传学特征,再根据这些特征进行分类。本研究中,通过对肿瘤标本进行相关检测,更倾向于将其归类为CIC重排肉瘤。
CIC-DUX4融合肉瘤的侵袭性较强,其5年生存率仅为14%~43% ,转移率高达44%~70%,主要转移部位是肺部[2]。本例患者术前行肺部CT和脊椎MRI检查均未发现转移病灶。该患者的病情进展较快,自出现右侧肢体麻木、无力至呈现嗜睡状态约半个月,经静脉滴注20%甘露醇后症状缓解,说明肿瘤生长速度快、并发水肿严重或存在肿瘤卒中的可能。该患者入院3 d病情即迅速恶化,急诊行头颅MRI检查发现病变增大后立即行手术切除,术中发现肿瘤实质部分出血,即肿瘤卒中。对其的手术治疗可参照侧脑室常见肿瘤如中枢神经细胞瘤、室管膜瘤等的治疗原则,尽量完整切除肿瘤。根据对本例患者的治疗经验,由于肿瘤生长速度极快,容易发生肿瘤卒中从而导致病情迅速恶化,今后对于类似病例应尽快完善相关检查,尽早手术,且尽可能全切除肿瘤。
目前,对于该肿瘤的治疗与尤因肉瘤类似,但治疗效果欠佳,有待进一步研究以发现有效的治疗方法[2,9]。尤因肉瘤家族的治疗主要以肿瘤的手术切除为主,辅以化疗及放疗等综合性治疗[3,10]。本例患者术后恢复良好,转外院行放射治疗(具体放疗方案不详)。目前术后随访3个月,患者的一般情况良好,恢复满意。
综上所述,CIC重排肉瘤分型较多,其中原发于中枢神经系统的CIC-NUTM1融合肉瘤罕见。明确诊断往往需要结合临床特点、形态学特征、免疫组织化学表型尤其是二代测序等分子检测结果。对于CIC重排肉瘤的治疗,除了强调手术完整切除、术后放疗等外,尚需寻找相应的靶向药物,并密切随诊观察。对于CIC重排肉瘤的生物学行为和临床转归,今后仍需更多、更大样本的研究明确。
所有作者声明无利益冲突

























