
探讨遗传性球形红细胞增多症(HS)患者的临床特点、基因突变检查结果及诊疗方法,并且进行相关文献复习。
选择2021年10月24日重庆医科大学附属儿童医院血液肿瘤科收治的1例8岁3个月男性HS合并室间隔缺损患儿为研究对象。采用回顾性分析方法,收集患儿病史、家族史资料,以及实验室及影像学检查结果,对患儿的临床表现、基因突变与诊疗过程进行分析。对患儿的随访截至2022年4月30日。进一步采集患儿及其父母外周血样进行基因测序,对检出的SLC4A1突变采用SIFT、PolyPhen-2、REVEL等遗传变异有害性预测工具评估蛋白结构功能的保守性。利用Swiss Model在线工具分析预测突变对于蛋白质结构的影响。以"遗传性球形红细胞增多症""SLC4A1基因""hereditary spherocytosis""SLC4A1"为中、英文关键词,在中国知网数据库、万方数据知识服务平台及PubMed数据库中检索HS相关文献,并对文献报道的HS患者进行分析和总结。文献检索时间设定为2019年1月1日至2022年4月29日。本研究获得重庆医科大学附属儿童医院伦理委员会审批(批准文号:2021.457),并与患儿家属签署临床研究知情同意书。
①本例患儿因"面色苍黄加重2个月+"就诊。患儿面色苍黄,病程中间断出现茶色尿,中度贫血,肝、脾大合并先天性室间隔缺损(膜周融合型)。血常规检查结果示,白细胞计数为7.06×109/L,红细胞计数为2.41×1012/L,血红蛋白(Hb)值为64 g/L,平均红细胞体积(MCV)为94.6 fL,平均血红蛋白浓度(MCHC)值为281 g/L,网织红细胞百分比为11.47%。外周血涂片检查结果示,患儿外周血球形红细胞比例为18.3%。生化全项检查结果示,总胆红素水平为53.1 μmol/L,直接胆红素水平为15.7 μmol/L,间接胆红素水平为37.4 μmol/L,乳酸脱氢酶水平为439 U/L。红细胞渗透脆性增加。基因测序结果示,患儿携带SLC4A1基因12号外显子c.1387G>A(p.Gly463Ser)罕见杂合错义突变,该突变来源于其父,预测为有害致病基因突变。患儿父亲未表现出贫血、溶血相关症状,仅外周血涂片可见少量球形红细胞。Swiss Model在线工具分析预测结果示,突变型SLC4A1蛋白的三级结构未因氨基酸替换受到影响。②根据患儿临床表现及基因检查结果,本例患儿被诊断为HS。患儿接受红细胞成分血输注及脾切除术,治疗后患儿临床症状基本消失,处于部分缓解状态。截至随访结束,患儿一般情况良好,拟择期行后续心室间隔缺损手术。③根据本研究设定的文献检索策略,共检索出5篇HS文献,报道6例HS患者,加上本研究1例,共纳入7例HS患者。其中,1例患者临床表现为皮肤黄染并远端肾小管酸中毒(dRTA),无HS家族史,SLC4A1错义突变致病。1例患者伴反复低血钾,外周血细胞形态学检查结果可见大量球形红细胞,有HS家族史,SLC4A1错义突变致病。1例患者伴反复不明原因贫血,有HS家族史,SLC4A1缺失突变致病。2例患者面色苍白、黄疸,SLC4A1新发错义突变致病。1例伴黄疸、胆石症,有HS家族史,SLC4A1错义突变致病。本例患儿面色苍黄,有HS家族史,SLC4A1错义突变致病。
本例HS患儿家系为SLC4A1基因12号外显子c.1387G>A(p.Gly463Ser)罕见杂合错义突变致病。基因突变检查有助于HS的确诊。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
遗传性球形红细胞增多症(hereditary spherocytosis,HS)是红细胞膜骨架蛋白异常引起的先天性慢性溶血性疾病[1]。目前暂无我国HS确切发病率的文献报道,估计为1.39/10万[2]。不同程度的贫血、黄疸和脾大为该病的主要临床表现,但是不同患者临床表现轻重不一,异质性强。各级临床医师对HS认识不足,导致该病诊治困难,误诊和漏诊率较高,甚至部分患者长期得不到确诊[3]。ANK1,SPTB,溶质载体家族4成员1(solute carrier family 4, member 1,SLC4A1),SPTA1及EPB42为目前已知的HS相关致病基因。其中,SLC4A1基因(OMIM:612653)在临床检出相对少见。本研究拟对1例伴SLC4A1罕见突变HS患儿的临床资料进行回顾性分析并检索相关文献,探讨HS致病基因突变的特点及相应诊断、治疗,从而提高临床工作者对HS的认识。现将研究结果报道如下。
选择2021年10月24日,重庆医科大学附属儿童医院收治的1例HS合并室间隔缺损患儿为研究对象。本例患儿为男性,8岁3个月。本研究获得重庆医科大学附属儿童医院伦理委员会审批(批准编号:2021.457),并与患儿家属签署临床研究知情同意书。
本例患儿进行的实验室检查包括血常规、骨髓细胞形态学、骨髓细胞免疫分型、血液生化、凝血及免疫相关检查等。影像学检查包括腹部超声、心脏超声等。本例患儿诊断、治疗、疗效评价均参照《血液病诊断及疗效标准》第4版[4]。
采集患儿及其父母的外周静脉血各2 mL,行基因测序筛查疾病相关突变位点,并采用Sanger测序法验证检出突变。基因突变检查委托康圣环球基因技术公司完成。
采用SIFT(http://sift.jcvi.org/)、PolyPhen-2(http://genetics.bwh.arvard.edu/pph2)及REVEL(https://sites.google.com/site/revelgenomics/)等遗传变异有害性预测工具评估蛋白结构功能的保守性。进一步利用Swiss Model在线工具(https://swissmodel.expasy.org/)对检出SLC4A1罕见突变进行突变对于蛋白质结构的影响预测。其中,SIFT分数范围0~1分,分值<0.05分被预测为有害;Polyphen2,基于HumanVar数据库,常用于单基因遗传病,预测结果为D(很可能有害,分值≥0.909分),P(可能有害,0.447分≤分值≤0.909分),B(无害,分值≤0.446分);REVEL分数范围为0~1分,分值>0.5分被预测为有害。
本例患者以门诊的方式对患者进行随访,门诊监测血常规以了解患儿血红蛋白(hemoglobin,Hb)值情况,随访频次为2~3个月1次。随访截至2022年4月30日。
本研究以"遗传性球形红细胞增多症""SLC4A1基因""hereditary spherocytosis""SLC4A1"为中、英文关键词,在中国知网数据库、万方数据知识服务平台及PubMed数据库中检索HS相关文献,通过阅读文献题目与摘要排除无关文献。检索时间设定为2019年1月1日至2022年4月29日。纳入包含基因检测结果及临床记录完整的HS病例。
本例患儿,苗族,籍贯贵州省。因"面色苍黄加重2个月+",2021年10月24日就诊于重庆医科大学附属儿童医院血液肿瘤科。患儿自幼间断面色发黄,2个月前无明显诱因出现面色苍黄加重,偶伴乏力。1个月前曾出现非喷射样呕吐伴纳差,无头痛、惊厥发作,未发现巩膜黄疸,无出血表现。病程中患儿间断出现茶色尿,晨起尿呈红茶色,中午至晚上逐渐转为啤酒色,可自行缓解,共出现3次,持续4 d。患儿于2021年9月3日外院就诊,血常规检查结果示,白细胞计数(white blood cell count,WBC)为2.64×109/L,红细胞计数为1.73×1012/L,Hb值为43 g/L,血小板计数为59.0×109/L。患儿行骨髓穿刺,骨髓细胞形态学检查结果示,增生性贫血骨髓象,巨核细胞增生活跃伴成熟不良。予输注去白细胞悬浮红细胞纠正贫血,并予口服泼尼松,治疗10 d,患儿面色无明显好转且仍间断解茶色尿,为进一步诊疗于本院就诊。患儿父母非近亲结婚,否认相关血液系统疾病家族史。
患儿入院后查体:体温为36.5 ℃,呼吸频次为21次/min,心率为118次/min,血压为105 mmHg/60 mmHg(1 mmHg=0.133 Pa),面罩吸氧下氧饱和度为97%,体重为23 kg,身高为115 cm。神清,表达清楚,贫血貌,双侧睑结膜苍白,巩膜及皮肤轻、中度黄染,腹软,肝肋下2 cm,剑突下4 cm,质软、无触痛,脾Ⅰ线7 cm、Ⅱ线7 cm、Ⅲ线5 cm,质偏中。实验室检查结果如下。血常规检查结果示,WBC为7.06×109/L,红细胞计数为2.41×1012/L,Hb值为64 g/L,平均红细胞体积(mean corpuscular volume, MCV)为94.6 fL,平均血红蛋白浓度(mean corpuscular Hb concentration, MCHC)为281 g/L,网织红细胞百分比为11.47%,血小板计数为66×109/L。血气分析结果示,pH值为7.48,PCO2为32 mmHg,PO2为249 mmHg,标准碱剩余为0.40 mmol/L。外周血涂片检查结果示,患儿外周血球形红细胞比例为18.3%;患儿父亲外周血球形红细胞比例为2.1%;患儿母亲外周血球形红细胞未检出。患儿尿常规检查结果示,尿胆原为35 μmol/L,其余指标未见异常。生化全项检查结果示,总胆红素水平为53.1 μmol/L,直接胆红素水平为15.7 μmol/L,间接胆红素水平为37.4 μmol/L,乳酸脱氢酶水平为439 U/L,其余指标基本正常。红细胞渗透脆性试验结果示,开始溶血时,盐水浓度为5.2 g/L(参考范围为3.8~4.6 g/L),完全溶血时,盐水浓度为为3.6 g/L(参考范围为2.8~3.2 g/L)。直接、间接抗人球蛋白试验结果均为阴性。葡萄糖-6-磷酸脱氢酶(glucose 6 phosphatedehydrogenase,G6PD)活性测定检查结果示,活性正常;凝血五项、心肌标志物+脑钠肽、大便常规、自身抗体全套、肝炎标志物检查等均未见明显异常。腹部超声检查结果示,肝大,肝右锁骨中线肋缘下4.0 cm,肝右叶斜径12.0 cm;脾大,脾左肋缘下8.3 cm,脾长径为14.2 cm,厚约为4.4 cm。心脏超声检查结果示,室间隔缺损(膜周融合型)为12 mm,伴重度肺动脉高压,三尖瓣反流(中度),右室腔异常粗大肌束,心包积液(少量)。根据患儿临床表现结合实验室检查、影像学检查结果,本例患儿初步诊断为慢性溶血性贫血,不伴代谢性酸中毒及电解质紊乱,HS可能性大。
基因检测结果示,本例患儿为先证者,携带SLC4A1基因12号外显子c.1387G>A(p.Gly463Ser)(NM_000342)罕见杂合错义突变,其父亲携带同样位点突变,其母亲该位点未见异常(图1)。经SIFT、PolyPhen-2、REVEL等变异有害性预测工具评估蛋白结构功能的保守性,患者SLC4A1罕见错义突变c.1387G>A在线软件预测其致病有害性结果分别为SIFT=0、PolyPhen-2=0.988、REVEL=0.759,上述预测结果均提示突变有害的可能性大。在不同物种间分析该突变位点的保守性,见图2。Swiss Model在线工具预测SLC4A1基因c.1387G>A(p.Gly463Ser)突变对于蛋白质结构影响的结果显示,该罕见错义突变导致第463位甘氨酸被丝氨酸取代,但是与野生型SLC4A1蛋白相比,蛋白的三级结构未因氨基酸替换受到影响(图3)。
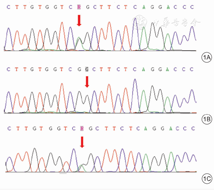
注:箭头示突变位点

注:红色箭头表示突变氨基酸,蓝色代表保守氨基酸,灰色代表较保守的氨基酸,白色代表不保守氨基酸。Homo sapiens为人;Mus musculus为小鼠;Rattus norvegicus为大鼠;Gallus gallus为鸡;Bos taurus为牛;Macaca mulatta为恒河猴
注:箭头示氨基酸位点改变
根据患儿临床表现及基因检查结果,本例患儿确诊为HS合并室间隔缺损(膜周融合型)。10月25日,患儿血常规检查结果示危急值,Hb值为47 g/L,立即给予悬浮红细胞及浓缩红细胞输注治疗。期间共输注悬浮红细胞8 U,浓缩红细胞2 U。碱化尿液、红细胞制剂输注等对症治疗措施使得患者溶血危象改善,无出血倾向。患儿于11月5日转入肝胆外科,于11月8日行脾切除术,11月16日患儿无发热,无咳嗽、咳痰,无恶心、呕吐,查体:神清,反应好,皮肤巩膜轻度黄染,双肺无罗音,心音有力律齐,未及杂音。腹平坦,未见肠型、蠕动波,腹软,无压痛,无反跳痛、肌紧张,肝肋下未及,叩诊鼓音,移动性浊音阴性,肠鸣音正常,手术切口愈合好,无需拆线,予出院。根据方法中的疗效评价标准,本例患儿临床症状基本消失,处于部分缓解状态。截至随访结束,患儿一般情况良好,拟择期行心室间隔缺损手术。
根据本研究设定的文献检索策略,共检索出5篇HS相关文献,其中中文文献2篇,英文文献3篇,共报道6例HS患者[5,6,7,8,9]。加上本例患儿共7例患者的基因型与临床表现总结,见表1。其中1例患儿临床表现为皮肤黄染并远端肾小管酸中毒(distal renal tubular acidosis,dRTA),无HS家族史,为SLC4A1错义突变致病。1例患者伴反复低血钾,外周血细胞形态学检查结果可见大量球形红细胞,有HS家族史,为SLC4A1错义突变致病。1例患者伴反复不明原因贫血,有HS家族史,为SLC4A1缺失突变致病。2例患者面色苍白、黄疸,为SLC4A1新发错义突变致病,先证者父母基因检测结果均未见异常。1例伴黄疸、胆石症,有HS家族史,为SLC4A1错义突变致病。本例患儿面色苍黄,有HS家族史,为SLC4A1错义突变致病。
7例伴SLC4A1基因突变HS患者的基因型与临床表现
7例伴SLC4A1基因突变HS患者的基因型与临床表现
| 文献(第一作者,发表年) | 性别 | 年龄 | 临床表现 | HS家族史/输血史 | 基因突变 | 突变类型 | 是否接受脾切除术 | 诊断 |
|---|---|---|---|---|---|---|---|---|
| 陈雁[5],2020 | 男 | 1岁7个月 | 乏力、食欲减退 | 无/有 | c.2102G>A(p.Gly701Asp)、c.1188T>C(p.Met663Thr) | 错义突变 | 否 | HS合并dRTA |
| Indra[6],2020 | 男 | 57岁 | 反复低钾 | 有/- | c.1478G>T(p.Trp496Leu) | 错义突变 | — | HS |
| Elias[7],2021 | 女 | 37岁 | 贫血 | 有/有 | c.2655+2_2655+3del | 缺失突变 | — | HS |
| 张勇刚[8],2019 | 男 | 5岁 | 面色苍白、黄疸 | 无/有 | c.37G>A(p.Gln13Lys) | 错义突变 | 否 | HS |
| 张勇刚[8],2019 | 女 | 3岁7个月 | 面色苍白、黄疸 | 无/有 | c.340T>G(p.Phe114Leu) | 错义突变 | 否 | HS |
| Shen[9],2019 | 女 | 24岁 | 黄疸、胆石症 | 有/有 | c.1469G>A(p.Arg490His) | 错义突变 | 是 | HS |
| 本研究 | 男 | 8岁3个月 | 面色苍黄 | 有/有 | c.1387G>A(p.Gly463Ser) | 错义突变 | 是 | HS合并室间隔缺损 |
注:"—"表示文献未提及相关数据。SLC4A1为溶质载体家族4成员1,HS为遗传性球形红细胞增多症,dRTA为远端肾小管酸中毒
HS是一种遗传性疾病,主要为常染色体显性遗传和隐性遗传,部分患者可携带新发突变,具有遗传学异质性[10]。ANK1、SPTB、SLC4A1、SPTA1和EPB42为目前已知的HS相关致病基因,上述基因分别编码红细胞膜骨架蛋白中的锚蛋白、β-血影蛋白、α-血影蛋白、带3蛋白及4.2蛋白[11]。中国人群HS具有独特的基因突变谱,其中SPTB突变率为45%,ANK1突变率为45%,SLC4A1突变率为10%,且突变均表现为非再现性及家系特异性[12]。编码红细胞膜相关蛋白的基因发生突变,导致细胞膜缺陷,红细胞独特的变形能力和机械稳定性因基因突变而丧失,最终被脾巨噬细胞吞噬,导致溶血发生为HS的发病机制[13]。因此,明确致病基因的突变位点对于研究HS分子生物学机制十分重要。本研究报道1例伴SLC4A1罕见突变HS患儿,对HS的诊断、治疗具有积极意义,并且进一步丰富了HS基因突变库[14]。
SLC4A1定位于17号染色体q21-q22,长度约为18 kb,包含20个外显子,该基因可同时编码2种阴离子交换蛋白,分别是位于红细胞表面的红细胞阴离子交换蛋白(erythrocyte anion exchange protein,eAE)1和位于肾闰细胞基膜侧的肾脏阴离子交换蛋白(kidney anion exchange protein,kAE)1,发挥维持红细胞膜稳定性及调节肾酸碱平衡的重要作用[15]。通过对比ClinVar数据库(https://www.ncbi.nlm.nih.gov/clinvar/?term=SLC4A1[gene])发现本例HS患儿SLC4A1基因c.1387G>A未见既往文献报道。该突变导致463位甘氨酸被丝氨酸取代,虽然经Swiss Model在线工具评估SLC4A1蛋白的三级结构没有因氨基酸替换受到影响,但是亲水基团替换疏水基团可能影响蛋白功能,导致疾病发生,但还需要进一步实验证实。此外,本例患儿及其父亲均存在同样的基因突变位点,但其父亲并未有任何临床表现,仅外周血涂片见少量球形红细胞。由此,笔者推测SLC4A1基因c.1387G>A错义突变为非功能缺失型突变,该突变仅导致无症状的HS,在存在诱因及合并症的情况下可发生急性溶血。
针对本研究报道的HS患儿病程中间断出现茶色尿的临床表现,可能是由于患儿先天性室间隔缺损(膜周融合型)的病情加重诱发患儿溶血失代偿发生溶血危象。文献报道,先天性心脏病患者若发生急性溶血通常发生在介入治疗后,高速血流冲击金属材质的介入器材,使红细胞受到撞击被破坏而发生机械性溶血,表现为排出洗肉水样或酱油色的Hb尿[16]。本例患儿为合并先天性心脏病的HS在没有任何临床干预的情况以急性溶血为首发表现就诊。对于同时具有血管内溶血和血管外溶血临床表现的HS患者诊断具有借鉴意义。1887年,脾切除术被首次应用于治疗HS,该方法被认为是治疗HS最有效的方法[17]。目前,尚无HS合并室间隔缺损发生急性溶血危象的患儿行脾切除手术治疗的相关报道。本例患儿出现溶血危象,室间隔缺损进一步加剧溶血。此时溶血危象、室间隔缺损、脾功能亢进的处置先后问题需要做出抉择。急性溶血期通过血液制品输注改善患者一般情况后行脾切除术,再行室间隔缺损手术为本院多学科会诊结果,且本例患儿病情控制情况好,值得借鉴。值得注意的是,仍有部分HS患儿,在接受脾切除术后依然表现为输血依赖[18]。长期输血导致的铁过载,以及脾切除术后对患儿免疫系统造成的损伤是常规治疗难以避免的不良反应[19,20]。随着相关研究进展,基因治疗为遗传性疾病提供了新治疗方案。文献报道,基因编辑介导的基因治疗将有望根治多种遗传性血液系统疾病[21]。采用成簇规律间隔的短回文重复序列(clustered regularly interspaced short palindromic repeat sequences,CRISPR)-CRISPR相关蛋白(CRISPR-associated protein,Cas)9技术治疗镰状细胞贫血、输血依赖型β-地中海贫血等疾病正在Ⅰ/Ⅱ期临床试验中[21]。而HS为编码红细胞膜蛋白的基因突变致病,基因治疗是其根治疗法[22],但具体疗效有待进一步研究证实。
HS患者多数为散发性就诊,误诊、漏诊在临床工作中较为常见。文献报道,1例病史达37年的轻症HS患者。可见,提高临床医师对HS的认识及诊断水平十分迫切[23]。血常规联合外周血涂片检查、红细胞渗透脆性试验、酸化甘油溶血试验、伊红-马来酰亚胺结合试验、十二烷基硫酸钠-聚丙烯酰胺凝胶电泳法及基因测序技术是目前临床较常用的HS辅助诊断方法[24]。其中,基因测序不仅可直接明确疾病诊断,还可以明确疾病的类别和分子生物学分型,提高诊断效率[25],具有广阔的应用前景。
综上所述,本研究报道1种SLC4A1罕见错义突变,为HS患者的诊断及遗传咨询提供指导,为进一步探索适合中国HS患儿致病基因的筛查诊断方法及HS发病机制提供了参考。关于手术时机的选择,HS合并室间隔缺损患儿出现溶血危象时,一般情况得到控制后及时行脾切除术可使患儿获益。基因治疗在临床应用中的不断探索与发展,为治愈HS患儿带来新的希望。
所有作者声明无利益冲突

























