
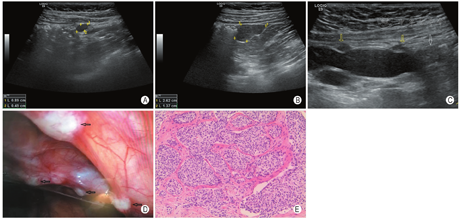
病例1,男,8岁5个月,因间歇性腹部钝痛2个月余来院就诊,超声检查发现腹腔及盆腔数处不规则低回声包块,最大者位于盆腔,长径4.8 cm;包块形态不规则,内部可见钙化斑,较大包块中央有少量液化,包块可见较丰富血流信号;肠间隙有少量清亮游离积液。对肿块进行手术切除,术后病理显示肿瘤细胞呈小圆形、巢状分布,核分裂象多见,周围见丰富纤维间隔(图1)。免疫组化:D99+、Desmin+、CK+、EMA+、WT1散在+、SYN-、MyoD1-、Desmin+、Ki-67约50%。基因检测显示EWSR1(22q12)基因断裂。病理诊断为促结缔组织增生性小圆细胞肿瘤。术后规律化疗,每隔1个月左右超声定期随访。4个月后超声检查发现腹膜复发灶,随访中逐渐增大(图1A,B)、增多,累及多个组织器官。行第二次手术,术中可见肿瘤弥漫种植于腹腔及盆腔的腹膜、网膜、肠系膜表面(图1D)及肠壁,膀胱壁浆膜侧,并在肝脏、脾脏等脏器内发现多发病灶,切除所有肉眼可见病灶,继续化疗。术后8个月超声检查再次发现复发病灶,随访中逐渐进展,间隔11个月后再次手术,切除肉眼可见病灶,术后5个月后多组织脏器再次出现多发复发病灶,并迅速增大,部分病灶逐渐包绕肠管、侵犯输尿管(图1C)。发病2年4个月后患儿死亡。
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
病例1,男,8岁5个月,因间歇性腹部钝痛2个月余来院就诊,超声检查发现腹腔及盆腔数处不规则低回声包块,最大者位于盆腔,长径4.8 cm;包块形态不规则,内部可见钙化斑,较大包块中央有少量液化,包块可见较丰富血流信号;肠间隙有少量清亮游离积液。对肿块进行手术切除,术后病理显示肿瘤细胞呈小圆形、巢状分布,核分裂象多见,周围见丰富纤维间隔(图1)。免疫组化:D99+、Desmin+、CK+、EMA+、WT1散在+、SYN-、MyoD1-、Desmin+、Ki-67约50%。基因检测显示EWSR1(22q12)基因断裂。病理诊断为促结缔组织增生性小圆细胞肿瘤。术后规律化疗,每隔1个月左右超声定期随访。4个月后超声检查发现腹膜复发灶,随访中逐渐增大(图1A,B)、增多,累及多个组织器官。行第二次手术,术中可见肿瘤弥漫种植于腹腔及盆腔的腹膜、网膜、肠系膜表面(图1D)及肠壁,膀胱壁浆膜侧,并在肝脏、脾脏等脏器内发现多发病灶,切除所有肉眼可见病灶,继续化疗。术后8个月超声检查再次发现复发病灶,随访中逐渐进展,间隔11个月后再次手术,切除肉眼可见病灶,术后5个月后多组织脏器再次出现多发复发病灶,并迅速增大,部分病灶逐渐包绕肠管、侵犯输尿管(图1C)。发病2年4个月后患儿死亡。
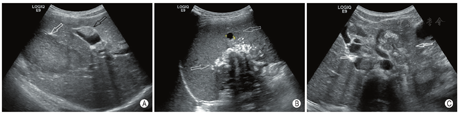
病例2,男,12岁1个月,因间歇性腹痛5个月余,在外院就诊时发现腹部包块来院。超声检查发现后腹膜、肝脏、胆囊壁、脾脏、膈肌等组织器官多发低回声包块,形态不规则,包块内可见多发沙砾样钙化斑(图2A, B),最大包块位于腹腔,长径16.0 cm;较小包块呈类椭圆形,较大包块呈分叶状,后腹膜部分包块互相融合包绕腹部大血管(图2C),包块内部血供丰富,较大包块可见液化区。行手术活检,术后病理诊断促结缔组织增生性小圆细胞肿瘤,基因检测显示EWSR1(22q12)基因断裂。活检后开始规律化疗,超声每隔1个月定期随访。化疗期间病灶增大、病变进展,发病1年2个月后患儿死亡。
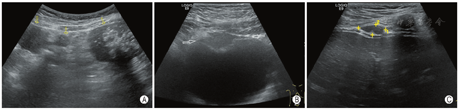
病例3,男,11岁5个月,因偶然发现腹部包块1个月余,在当地医院活检术后病理考虑促结缔组织增生性小圆细胞来院就诊。超声检查发现腹腔及盆腔肠系膜、大网膜、腹膜、膈肌、肝脏,肠壁、膀胱壁浆膜侧等处可见多发大小不等低回声包块,形态不规则、呈分叶状,较大病灶内多发砂砾样钙化灶,部分可见声影(图3),肠间隙有少量清亮腹腔积液。规律化疗后行手术切除,切除肉眼可见所有病灶,术后病理诊断促结缔组织增生性小圆细胞肿瘤,基因检测显示EWSR1基因断裂阴性。术后继续化疗并每隔1个月左右定期超声随访,术后2个月后超声显示膀胱壁复发病灶,目前仍在化疗中,肿瘤有所缩小。该病例发病至今随访时长共7个月。
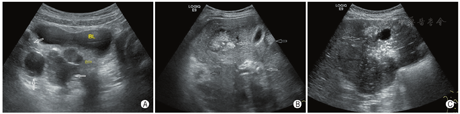
病例4,男,10岁3个月,因间歇性腹痛3周,当地医院发现腹部多发占位,行穿刺活检,术后病理考虑促结缔组织增生性小圆细胞瘤肿瘤来诊,基因检测显示EWSR1(22q12)基因断裂。超声检查可见盆腔、腹腔肠系膜、大网膜、肠壁、肝脏、肝包膜、脾脏、膈肌、膀胱壁、肠壁、左肾、后腹膜等部位多发大小不等低回声包块,形态不规则,表面分叶状,较大包块可见多发钙化斑及液化区,部分包块互相融合,包块最大者位于肝左叶,长径18.3 cm,凸向肝外生长(图4)。胸部CT提示右肺胸膜多发块状、结节状软组织密度影,考虑肿瘤转移灶。来院后开始规律化疗,每隔1个月定期超声随访,目前共随访3个月,肿瘤有所缩小。
本研究通过浙江大学医学院附属儿童医院伦理委员会审查批准(批件号2022-IRB-139)。
促结缔组织增生性小圆细胞肿瘤(desmoplastic small round cell tumor,DSRCT)是一种组织起源未定的罕见肿瘤,发病率为(0.1~0.4)/1 000 000[1,2]。DSRCT无特异性临床表现,早期诊断困难,该疾病恶性程度极高,5年生存率仅为15%[3]。DSRCT好发于青少年,确诊时年龄18~22岁[4],以男性多见,男女比例约为4∶1[5]。本研究中病例年龄较以往报道低,但在儿童中均属于较大年龄段的学龄期儿童。尽管也有研究认为小于20岁病例中女性比例高于男性[6],但本研究患者均为男性。DSRCT确诊主要依靠病理诊断和分子基因检测,病理主要表现为:小圆细胞肿瘤巢状分布,可见核仁和核分裂象,周围大量纤维结缔组织增生,免疫组化可表达上皮性、肌性和神经性分化相关蛋白,大多数病例CK、EMA、vimentin、desmin和NSE阳性。90%病例分子病理学有特异性的22q12上的EWSR1基因断裂,并与11p13上的WT1基因融合[7,8],本组病例均得到病理证实,同时3例有特征性分子病理学表现。
DSRCT好发于腹腔和盆腔,肿瘤多位于腹盆腔和脏器浆膜面,容易侵犯周围组织器官并远处转移[9,10,11],有研究表明仅20%病例发病时病变局限[6]。本研究所有病例就诊时均在腹腔和盆腔发现多发病灶,分布于多个组织器官。若参照基于影像学表现对DSRCT危险分层的研究[12],将同时有肝转移和腹水为极高风险,有肝转移或腹水为高风险,两者皆无为中风险,本组病例2例为极高风险,2例为高风险,无中风险病例。表明小儿DSRCT起病隐匿,早期发现困难。
DSRCT侵袭性高,易复发,预后差,中位生存期17.8~20个月[2],70%病例在确诊后3年内死亡[13]。DSRCT目前无标准治疗模式,治疗以手术、化疗、放疗等综合治疗方案为主[1,3,9],近年来也有免疫疗法的报道[4],但治疗效果仍然不理想。即使多模态综合治疗,DSRCT的无进展生存期仅为3.9个月[1]。本组所有病例术前、术后均进行规律化疗,并有2例进行了手术切除。其中有1例(例1)术后反复复发,超声能及时发现并观察复发灶大小、数目变化,随疾病进展共进行了3次手术;1例(例3)术中肉眼可见病灶切除后2个月出现复发;而另1例患儿大量病灶弥漫分布,在化疗期间病变进展并死亡。以上表现均符合该疾病无进展生存期短的特点。
目前中英文文献仅有少量的DSRCT影像学研究,并认为CT是首选的检查方式,MRI对腹盆腔疾病有帮助[10,14],DSRCT的超声研究鲜有报道。本研究显示,DSRCT在超声上表现为腹腔或盆腔大小不等的低回声包块,病灶数量众多,可弥漫分布,并累及多部位、多器官,但病灶大小、分布无规律性,因此难以定位原发灶来源。包块以低回声为主,内部可见多发沙砾样钙化斑,较大包块有液化坏死区。病灶多见于腹膜、大网膜、肠系膜、膈肌等器官表面以及肠壁、膀胱壁浆膜面,也可见于输尿管管壁。实质脏器中肝脏、脾脏受累多见,胰腺、肾脏也可受累。小儿的腹、盆腔病变有以上超声表现时,应考虑到DSRCT。
在术后随访中,本研究显示超声可及时发现复发病灶,复发灶的形态、回声与术前一致。在定期监测中,可见病灶逐渐增多、增大,直至侵犯、压迫相应组织器官,也可包绕肠道造成梗阻。超声在随访监测中可为临床治疗决策提供重要信息。
DSRCT包块的回声及钙化与儿童肿瘤中较多见的神经母细胞瘤有相似之处,神经母细胞瘤以婴幼儿及学龄前儿童多见,多位于脊柱旁的后腹膜、后纵隔以及肾上腺,肿瘤形态大多呈多结节状;较大肿瘤易包绕邻近腹部大血管,液化坏死较少见[15]。肝脏内包块也需与肝母细胞瘤鉴别,肝母细胞瘤内可有少量细小钙化和小囊状无回声,肝母细胞瘤一般位于肝实质内为主,转移器官以肺多见,少有多部位组织、器官多发、弥漫分布的表现。小儿腹腔、盆腔肿瘤中横纹肌肉瘤也较多见,但横纹肌肉瘤一般无钙化,液化坏死区多见,尽管侵袭性较强,但多器官大量大小不等病灶分布则少见。
综上所述,小儿DSRCT多见于学龄期儿童,超声表现有一定的特征性并在随访检查中可发挥重要作用。
所有作者均声明不存在利益冲突

























