总结新生儿遗传性癫痫的致病基因和临床特点。
选择2016年7月至2021年5月北京儿童医院新生儿中心收治并进行癫痫基因测序分析且结果异常的遗传性癫痫患儿进行回顾性研究,根据基因变异类型分为离子通道变异组和非离子通道变异组,比较两组患儿临床特点、治疗及预后情况。
共纳入36例,涉及15个致病基因,致病基因前3位为KCNQ2、SCN2A、STXBP1。离子通道变异组20例(55.6%),非离子通道变异组16例(44.4%),两组一般情况、发作类型、脑电图特点、治疗及转归比较,差异均无统计学意义(P>0.05)。36例患儿中,癫痫起病日龄为生后10 min至24 d,生后1周内发病28例(78.8%)。诊断发育性癫痫性脑病20例(55.6%),自限性新生儿癫痫7例(19.4%),吡哆醇依赖症、Zellweger综合征各2例(5.6%),自限性家族性新生儿婴幼儿癫痫、Turner 型智力低下并癫痫、PURA综合征、Rett综合征、22q11.2缺失综合征各1例(2.8%)。使用的抗癫痫药物包括苯巴比妥、左乙拉西坦、奥卡西平、托吡酯、丙戊酸钠、苯二氮卓类、拉考沙胺、拉莫三嗪共8种。5例患儿放弃治疗后死亡,31例随访了6~50个月,22例(71.0%)发作控制,控制年龄为1月余至35个月,其中21例(56.7%)存在发育落后;6例(19.4%)控制无效,3例(9.7%)发作减少,均有不同程度发育落后。
新生儿期起病的癫痫与多种基因变异相关,KCNQ2、SCN2A、STXBP1是常见致病基因,其中KCNQ2 基因变异最多,多数患儿在生后1周内起病。离子通道相关基因变异占半数以上,钠离子通道阻滞剂有一定疗效。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
新生儿癫痫发作是新生儿神经系统常见急症,发病率为(1~5)/1 000活产儿[1]。有研究表明13%的新生儿惊厥发作会发展为新生儿癫痫[2]。2021年国际抗癫痫联盟(the International League Against Epilepsy,ILAE)将新生儿癫痫病因分为六大类,包括新生儿缺氧缺血性脑病(hypoxic-ischemic encephalopathy,HIE)、结构性、代谢性、遗传性、感染性和病因未明性[1]。除HIE、缺血性或出血性脑卒中或急性感染性病因外,新生儿癫痫常由潜在遗传因素导致,新生儿癫痫病因中60%与遗传性因素有关[3]。随着二代测序技术在临床中的应用,越来越多的癫痫致病基因被发现。这些基因涉及离子通道、细胞信号、DNA修复、代谢途径等[4]。本研究对2016年7月至2021年5月本中心收治的新生儿期起病的遗传性癫痫患儿的致病基因和表型特点进行研究,为临床决策和指导治疗提供依据。
选择2016年7月至2021年5月北京儿童医院新生儿中心收治的新生儿癫痫患儿进行回顾性分析。入选标准:(1)生后28 d内发病;(2)符合新生儿癫痫诊断;(3)住院期间完成头颅影像学和脑电图检查,且临床资料完整。排除标准:(1)病因为获得性脑损伤(如HIE、低血糖脑损伤、Ⅱ级以上颅内出血、脑梗死、中枢神经系统感染等);(2)未行基因检测,或基因检测未见异常、变异致病性不明确;(3)存活患儿随访时间少于3个月。根据基因变异情况分为离子通道变异组和非离子通道变异组。本研究已通过首都医科大学附属北京儿童医院医学伦理委员会审批(2022-E-194-R)。
1.临床资料:通过查阅电子病历,记录患儿性别、胎龄、癫痫起病日龄、发作类型、围产期情况、个人史、家族史、辅助检查结果(脑电图、头颅磁共振、遗传代谢病筛查等)及治疗情况。通过保健科及神经内科门诊复诊及电话随访预后,记录随访时间、发作情况、用药情况、发育情况(包括复诊时体格检查结果及家长电话描述)。
2.基因检测方法:采用靶向捕获二代测序癫痫基因检测包或全外显子组测序的方法明确患儿的致病基因,进一步采用Sanger测序验证相关变异位点并明确变异来源,并在家系中进行共分离分析,部分行拷贝数变异检查。应用Mutation Taster、SIFT和Polyphen-2进行蛋白质结构破坏和疾病诱发可能性分析。根据美国医学遗传学和基因组学学会指南评估变异的致病性[5]。
3.相关定义:(1)新生儿癫痫[1]:至少满足以下一项:①病程中至少2次无诱因癫痫发作,间隔>24 h;②1次无诱因发作,但与2次无诱因发作后再发风险相似(60%以上);③符合癫痫综合征诊断(如大田原合征等)。发作形式分为自动症、阵挛、强直、肌阵挛、癫痫性痉挛等。(2)根据2021年ILAE新生儿癫痫分类标准[6],将癫痫综合征分为自限性新生儿癫痫、自限性家族性新生儿婴幼儿癫痫、发育性癫痫性脑病。(3)抗癫痫药物治疗疗效判断:发作控制(至少3个月无发作)、发作减少(发作次数减少≥25%)、无效(发作次数减少<25%或加重)。
应用SPSS 23.0软件进行统计学分析。正态分布的计量资料以±s表示,两两比较采用最小显著性法(least-significant difference,LSD)t检验;非正态分布的计量资料以M(Q1,Q3)表示,组间比较采用秩和检验;计数资料以例(%)表示,组间比较采用χ2检验。P<0.05为差异有统计学意义。
研究期间共收治新生儿癫痫703例,排除获得性损伤536例、未行基因检测56例、基因检测无异常35例、基因检测结果致病性不明确33例、随访时间少于3个月7例,最终纳入36例。
36例基因诊断明确患儿中,男19例,女17例。癫痫发作起病时间为生后10 min至生后24 d,中位起病时间为生后1.5(0.6,4.8)d,其中28例(77.8%)生后1周内发病。29例出生史无明显异常,7例有生后缺氧窒息史,2例有硬膜下出血,6例有癫痫家族史。
36例患儿共涉及15个致病基因和1例大片段缺失。离子通道基因变异20例,包括KCNQ2基因变异15例(新发变异14例、遗传性变异1例)、SCN2A基因变异3例(均为新发变异)、KCNT1基因变异1例(新发变异)、GABRB3基因变异1例(遗传性变异);非离子通道变异16例,包括STXBP1基因变异3例(均为新发变异)、ALDH7A1基因变异2例(均为遗传性变异)、PEX1基因变异2例(均为遗传性变异),MECP2、HUWE1、FGF12、GNAO1、ARX、CDKL5、PURA、EFHC1基因变异各1例(均为新发变异),22q11.2缺失1例。
1.临床表现:(1)离子通道变异组:发作类型包括强直8例(7例KCNQ2,1例SCN2A),痉挛5例(KCNQ2、SCN2A各2例,1例GABRB3),自动症4例(2例KCNQ2,SCN2A、KCNT1各1例),阵挛3例(2例KCNQ2,1例SCN2A),肌阵挛3例(2例KCNQ2,1例GABRB3),有3例患儿出现2种发作类型,1例(SCN2A)由阵挛发展成痉挛,1例(SCN2A)由强直发展成痉挛,1例(GABRB3)由肌阵挛发展成痉挛。(2)非离子通道变异组:发作类型包括强直5例(MECP2、HUWE1、FGF12、PEX1、EFHC1各1例),痉挛4例(2例STXBP1,GNAO1、ARX各1例),自动症3例(STXBP1、ALDH7A1、22q11.2缺失各1例),阵挛2例(CDKL5、PEX1各1例),肌阵挛2例(ALDH7A1、PURA各1例)。两组性别、胎龄、出生体重、起病日龄、发作类型比较,差异均无统计学意义(P>0.05),见表1、2。
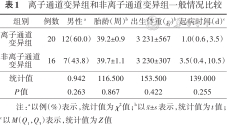
离子通道变异组和非离子通道变异组一般情况比较
离子通道变异组和非离子通道变异组一般情况比较
| 组别 | 例数 | 男性a | 胎龄(周)b | 出生体重(g)b | 起病时间(d)c |
|---|---|---|---|---|---|
| 离子通道变异组 | 20 | 12(60.0) | 39.2±0.9 | 3 231±567 | 1.0(0.6,3.5) |
| 非离子通道变异组 | 16 | 7(43.8) | 39.7±1.1 | 3 230±307 | 3.5(0.4,10.5) |
| 统计值 | 0.942 | 116.500 | 153.500 | 139.000 | |
| P值 | 0.263 | 0.867 | 0.422 | 0.255 |
注:a以例(%)表示,统计值为χ2值;b以±s表示,统计值为t值;c以M(Q1,Q3)表示,统计值为Z值
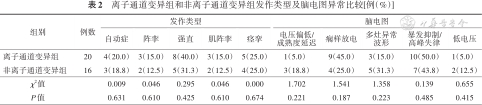
离子通道变异组和非离子通道变异组发作类型及脑电图异常比较[例(%)]
离子通道变异组和非离子通道变异组发作类型及脑电图异常比较[例(%)]
| 组别 | 例数 | 发作类型 | 脑电图 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 自动症 | 阵挛 | 强直 | 肌阵挛 | 痉挛 | 电压偏低/成熟度延迟 | 痫样放电 | 多灶异常波形 | 暴发抑制/高峰失律 | 低电压 | ||
| 离子通道变异组 | 20 | 4(20.0) | 3(15.0) | 8(40.0) | 3(15.0) | 5(25.0) | 1(5.0) | 9(45.0) | 3(15.0) | 10(50.0) | 1(5.0) |
| 非离子通道变异组 | 16 | 3(18.8) | 2(12.5) | 5(31.3) | 2(12.5) | 4(25.0) | 3(18.8) | 4(25.0) | 5(31.3) | 7(43.8) | 2(12.5) |
| χ2值 | 0.009 | 0.046 | 0.295 | 0.046 | 0.000 | 1.702 | 1.541 | 1.358 | 0.139 | 0.655 | |
| P值 | 0.631 | 0.610 | 0.425 | 0.610 | 0.674 | 0.221 | 0.187 | 0.223 | 0.485 | 0.415 | |
2.脑电图检查:(1)离子通道变异组:监测到暴发抑制或高峰失律7例(5例KCNQ2,SCN2A、GABRB3各1例),痫样放电6例(5例KCNQ2、1例SCN2A),暴发抑制及痫样放电3例(2例KCNQ2、1例SCN2A),多灶性放电2例(KCNQ2、KCNT1各1例),1例成熟度延迟、电压偏低(KCNQ2),1例暴发抑制及多灶样放电(SCN2A),1例低电压(KCNQ2)。(2)非离子通道变异组:监测到暴发抑制或高峰失律3例(2例STXBP1、1例HUWE1),成熟度延迟、电压偏低3例(ARX、PURA、大片段缺失各1例),痫样放电2例(均为PEX1),2例多灶性放电(CDKL5、EFHC1各1例),2例暴发抑制及痫样放电(STXBP1、GNAO1各1例),2例暴发抑制及多灶样放电(MECP2、FGF12各1例),1例低电压(ALDH7A1),1例多灶样放电及低电压(ALDH7A1)。见表2。
3.头颅影像检查:36例患儿中21例头颅磁共振成像结果异常,离子通道变异组11例(55.0%),包括脑实质异常信号7例,硬膜下出血2例,多微小脑回(GABRB3)、脑白质损伤各1例。非离子通道变异组10例(62.5%),脑实质异常信号5例,脑白质损伤2例、多微小脑回2例(均为PEX1)、脑回增宽1例(GANO1)。
4.诊断:36例患儿中,诊断发育性癫痫性脑病20例(55.6%),离子通道变异组13例,涉及KCNQ2基因9例,SCN2A基因2例,KCNT1、GABRB3基因各1例,其中KCNQ2、SCN2A、KCNT1均为新发变异;非离子通道变异组7例,涉及STXBP1基因3例,FGF12、GNAO1、ARX、CDKL5基因各1例。诊断自限性新生儿癫痫7例(19.4%),离子通道变异组6例,涉及KCNQ2基因5例,SCN2A基因1例,非离子通道组1例,为EFHC1基因;吡哆醇依赖症、Zellweger 综合征各2例(5.6%);自限性家族性新生儿婴幼儿癫痫1例(2.8%),为KCNQ2基因;Turner 型智力低下并癫痫、Rett 综合征、PURA 综合征、22q11.2缺失综合征各1例(2.8%)。
抗癫痫药物按应用情况排序为苯巴比妥(32例)、左乙拉西坦(13例)、奥卡西平(13例)、托吡酯(10例)、丙戊酸钠(8例)、苯二氮卓类(硝西泮/氯硝西泮/氯巴占/咪达唑仑,5例)、拉考沙胺(4例)、拉莫三嗪(1例)。1例(STXBP1)应用促皮质素治疗,短期内发作减少,后反复,联合生酮饮食治疗,发作减少>50%。1例(ALDH7A1)应用维生素B6治疗,发作控制。1例(22q11.2缺失)应用钙剂治疗,发作控制。应用单一抗癫痫药物治疗,离子通道组5例(25.0%),非离子通道组7例(43.8%);应用二联抗癫痫药物治疗,离子通道组3例(15.0%),非离子通道组4例(25.0%);应用三联及以上抗癫痫药物治疗,离子通道组12例(60.0%),非离子通道组5(31.3%)。见表3。
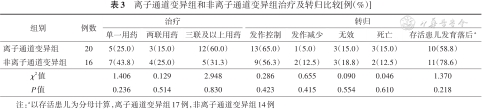
离子通道变异组和非离子通道变异组治疗及转归比较[例(%)]
离子通道变异组和非离子通道变异组治疗及转归比较[例(%)]
| 组别 | 例数 | 治疗 | 转归 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 单一用药 | 两联用药 | 三联及以上用药 | 发作控制 | 发作减少 | 无效 | 死亡 | 存活患儿发育落后a | ||
| 离子通道变异组 | 20 | 5(25.0) | 3(15.0) | 12(60.0) | 13(65.0) | 1(5.0) | 3(15.0) | 3(15.0) | 10(58.8) |
| 非离子通道变异组 | 16 | 7(43.8) | 4(25.0) | 5(31.3) | 9(56.3) | 2(12.5) | 3(18.8) | 2(12.5) | 11(78.6) |
| χ2值 | 1.406 | 0.129 | 2.948 | 0.286 | 0.655 | 0.090 | 0.046 | 1.370 | |
| P值 | 0.236 | 0.514 | 0.830 | 0.423 | 0.415 | 0.554 | 0.610 | 0.218 | |
注:a以存活患儿为分母计算,离子通道变异组17例,非离子通道变异组14例
离子通道变异组应用钠离子通道阻滞剂(sodium channel blockers,SCB)及发作控制情况:4例KCNQ2基因变异患儿(均为发育性癫痫性脑病)应用奥卡西平,3例控制,1例无效;1例SCN2A基因变异患儿(自限性新生儿癫痫)应用奥卡西平发作控制,1例SCN2A基因变异患儿(发育性癫痫性脑病)在3个月内应用奥卡西平联合拉考沙胺发作控制,另1例SCN2A基因变异患儿(发育性癫痫性脑病)应用拉考沙胺无效,1例GABRB3基因变异患儿联合应用奥卡西平、沙考拉安及拉莫三嗪无效。
36例中5例(13.9%)放弃治疗后在3个月内死亡,31例存活患儿随访6~50个月。
离子通道变异组20例患儿中,13例(65.0%)发作控制(11例KCNQ2,2例SCN2A),中位发作控制月龄为3.0(2.0,6.5)个月,其中6例(30.0%)存在智力运动发育落后(5例KCNQ2,1例SCN2A);1例(5.0%)发作减少(KCNT1);3例(15.0%)控制无效(KCNQ2、SCN2A、GABRB3各1例),均存在智力运动发育落后。
非离子通道变异组16例患儿中,9例(56.3%)发作控制(2例STXBP1,HUWE1、ARX、PURA、PEX1、EFHC1、ALDH7A1、22q11.2缺失各1例),中位发作控制月龄为6.0(4.8,15.0)个月,其中6例(37.5%)存在智力运动发育落后(2例STXBP1,HUWE1、ARX、PURA、PEX1各1例);2例(12.5%)发作减少(FGF12、CDKL5各1例);3例(18.6%)控制无效(STXBP1、MECP2、GNAO1各1例),均存在智力运动发育落后。
两组转归情况比较差异无统计学意义(P>0.05)。见表3。
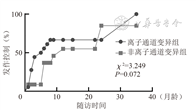
离子通道变异组与非离子通道变异组发作控制分别为13例(76.5%)、9例(64.3%),中位发作控制月龄分别为3.0(2.0,6.5)个月、6.0(4.8,15.0)个月,两组差异无统计学意义(P>0.05)。见图1。
新生儿遗传性癫痫起病早,寻找病因较困难,基因检测快速而准确,对病因不明的癫痫早期诊断和指导治疗有重大意义[7]。本研究共纳入36例新生儿期起病的癫痫患儿,起病最早为生后10 min,中位起病时间为生后1.5 d,多数(77.8%)生后1周内发病;本研究共涉及15个基因,前3位基因分别为KCNQ2(41.7%)、SCN2A(8.3%)和STXBP1(8.3%),与国内外报道一致[8, 9, 10]。遗传性癫痫病因包括离子通道相关基因变异和非离子通道相关基因变异,本研究中离子通道基因变异占55.6%,包括钾离子通道基因(KCNQ2、KCNT1)、钠离子通道基因(SCN2A)和γ-氨基丁酸(GABA)受体基因(GABRB3)。
离子通道基因表型谱广,具有表型异质性。本研究中有3例患儿出现2种发作类型,均为离子通道基因变异,20例离子通道基因变异患儿中,13例表现为发育性癫痫性脑病,6例为自限性新生儿癫痫,自限性家族性新生儿婴幼儿癫痫1例;其中发育性癫痫性脑病患儿中9例(9/13)为KCNQ2变异,均为新发变异,与文献报道一致[4]。SCN2A基因变异数量仅次于KCNQ2,SCN2A编码电压门控钠离子通道α2亚基,起病越早,功能增强型变异可能性越高[11],生后3个月内出现首次癫痫发作的患儿使用SCB后癫痫发作频率明显减少甚至完全控制;首次癫痫发作在出生3个月后的患儿使用SCB癫痫控制效果甚微[12]。本研究中2例(2/3)SCN2A基因变异癫痫表型为发育性癫痫性脑病,均为新发变异,1例在3个月内加用SCB,癫痫得到控制,而另1例未能及时应用SCB,发作未控制,与文献报道一致[12]。研究发现,电压门控钠离子通道与钾离子通道在细胞膜表面共存,KCNQ2功能增强型变异应用SCB可能有效[13]。本研究离子通道变异组中7例应用奥卡西平,5例发作控制,提示SCB对钠/钾离子通道变异者治疗有效。在临床中,新生儿期起病的不明原因癫痫患儿,怀疑遗传相关因素者,以KCNQ2、SCN2A等离子通道基因变异多见,可早期尝试SCB治疗,控制效果较好。本研究发现,与非离子通道变异组相比,离子通道变异组癫痫发作控制中位时间更短(3个月比6个月),在早期可能更容易控制,但随着随访时间延长,两组差异逐渐缩小。
非离子通道基因变异以STXBP1多见,在新生儿期不易控制癫痫发作,25%的患儿为难治性癫痫,既往文献报道中提到左乙拉西坦可以作为联合用药治疗STXBP1基因变异导致的癫痫,效果明显[14]。本研究中非离子通道变异组诊断发育性癫痫性脑病8例,其中3例为STXBP1基因变异,2例并未应用左乙拉西坦但发作得到控制,与文献报道并不一致,可进一步积累样本进行观察;3例STXBP1基因变异患儿随访均有智力运动发育明显落后,与既往文献[15]报道一致;另5例为HUWE1、FGF12、GNAO1、ARX、CDKL5基因变异,仅ARX基因变异患儿发作得到控制,4例均存在发育落后,与既往文献报道的癫痫性脑病一致[16, 17]。EFHC1基因变异既往认为和青少年肌阵挛性癫痫有关,也有文献报道EFHC1变异可引起严重的新生儿癫痫综合征[18]。本研究中EFHC1变异患儿经治疗后,6个月发作控制,无明显发育落后,与文献报道不一致,需继续随访至青少年期。本研究中有2例ALDH7A1基因变异,1例患儿加用维生素B6治疗,发作很快得到控制,无发育落后。ALDH7A1基因变异引起的吡哆醇依赖症在新生儿癫痫发作中并不少见[6],可实验性应用维生素B6治疗。另外,本研究中还有一部分先天综合征相关的基因变异,如HUWE1、MECP2、PEX1、PURA等基因变异,分别可导致Turner 型智力低下并癫痫、Rett 综合征、Zellweger 综合征、PURA 综合征等,均可出现新生儿期癫痫的表现,其中2例Zellweger 综合征患儿均合并多微小脑回畸形,与文献报道一致[18, 19, 20, 21]。另外本研究中有2例患儿合并脑结构畸形,分别为GNAO1和GABRB3基因变异,癫痫均未控制。
综上,新生儿遗传性癫痫多在生后1周内起病,早期行基因检测对诊断和治疗十分重要。新生儿期起病的癫痫与多个基因相关,离子通道相关基因变异占半数以上,早期尝试SCB有一定效果。
郑侠, 董世霄, 翁景文, 等. 新生儿遗传性癫痫36例临床分析[J]. 中华新生儿科杂志, 2023, 38(7): 401-406. DOI: 10.3760/cma.j.issn.2096-2932.2023.07.003.
所有作者声明无利益冲突

























