
研究巨结肠家系RET基因新发无义突变c.2599G>T是否具有致病功能。
利用定点突变技术构建RET c.2599G>T突变型过表达腺病毒,感染人胚肾细胞HEK-293。实验分为4组:未感染组(Control组)、阴性对照腺病毒对照组(NC组)、RET野生型过表达腺病毒组(WT组)、RET c.2599G>T突变型过表达腺病毒组(c.2599G>T组)。利用RT-PCR、蛋白质印迹法、免疫荧光定位、Transwell迁移实验、CCK-8增殖实验、流式细胞凋亡检测技术,胶质细胞源性神经营养因子(glial-derived neurotrophic factor,GDNF)处理细胞后检测p-RET和p-ERK1/2,探究突变RET基因的功能。多组均数间比较采用Tukey's T检验,多个研究组均数与对照组均数比较采用Dunnett-T检验。
c.2599G>T组不能检测到RET全长蛋白。Transwell检测细胞迁移个数c.2599G>T组(100.80±14.72)较WT组(155.20±7.89)迁移能力下降(P<0.001)。CCK-8检测细胞增殖能力c.2599G>T组比WT组分别在24 h (0.68±0.06比0.83±0.10)、48 h(0.90±0.10比1.17±0.13)、72 h(1.07±0.11比1.401±0.19)和96 h(1.38±0.12比1.68±0.15)明显下降(P<0.05)。流式细胞技术检测凋亡,c.2599G>T组3.12%较WT组0.85%增加(P<0.001)。应用GDNF处理细胞后,蛋白质印迹法检测p-RET和p-ERK1/2蛋白表达,c.2599G>T组(0.79±0.02,5.60±0.02)较WT组(72.04±0.58,10.72±0.02)均明显下降(P<0.001)。
RET基因无义突变c.2599G>T,是具有功能的新发突变,具有致病性,对先天性巨结肠的遗传咨询具有指导意义。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
先天性巨结肠(Hirschsprung disease,HSCR)是一种常见的先天性消化道畸形,其病理特征是在病变肠管位于肌层间的神经丛和黏膜下神经丛内的神经节细胞完全缺如。发病率为1/5 000至1/2 000,存在种族差异,亚洲人口发病率最高,本病有家族发生倾向[1,2]。目前研究认为HSCR是典型的多个基因共同参与的复杂人类遗传病,RET基因是HSCR的主要易感基因[3,4]。目前检测到的RET基因突变大多是通过测序分析得出的结果,是否具有致病功能,仍需进一步研究[5,6]。本课题组在1例巨结肠家系中应用二代测序并经Sanger法验证,发现RET基因14号外显子一个新的无义突变c.2599G>T,氨基酸改变为P.867 E>X (248),即第867位氨基酸变为终止氨基酸,引起RET蛋白截短248个氨基酸,14号外显子位于RET蛋白的酪氨酸激酶结构域,高度怀疑该突变是该家系的致病性突变[7]。细胞外调节蛋白激酶(extracellular regulated protein kinases,ERK)包括ERK1和ERK2,是RET的重要下游信号,磷酸化激活的ERK1/2由细胞质转位到细胞核内,参与细胞增殖与分化、细胞形态维持、细胞骨架的构建和细胞凋亡等多种生物学反应。在此基础上,本课题组通过体外实验对此新发突变进行了功能研究。
人胚肾细胞HEK-293(中国赛百慷生物技术有限公司),腺病毒、蛋白提取试剂盒、BCA蛋白浓度测定试剂盒、细胞凋亡检测试剂盒、CCK-8试剂盒(中国万类生物科技有限公司),RT-PCR反转录酶(中国碧云天生物技术公司),Matrigel胶、Transwell小室(美国Corning公司),Cy3标记山羊抗兔IgG(美国Invitrogen公司)。本研究经中国医科大学附属盛京医院伦理委员会审核通过(批准编号:2021PS194K)。
人胚肾细胞HEK-293采用含10%胎牛血清的MEM培养基于37 ℃、5%CO2的培养箱内常规培养。
构建人源RET(NM_020975.6,CDS区长度为3 345 nt)野生型过表达腺病毒及阴性对照腺病毒,利用定点突变技术构建RET c.2599G>T(p.867,E>X(248),即第867号氨基酸由谷氨酸突变为终止氨基酸致使RET蛋白缩短了248个氨基酸)突变型过表达腺病毒,带有血凝素标签。调整HEK-293细胞状态,选择对数生长期细胞进行感染。
实验分为未感染组(Control组)、阴性对照腺病毒对照组(NC组)、RET野生型过表达腺病毒组(WT组)、RET c.2599G>T突变型过表达腺病毒组(c.2599G>T组)。
细胞感染24 h后检测各组细胞中RET基因的表达。Trizol法提取细胞总RNA后,NANO 2000测定各样本中RNA的浓度。用反转录试剂盒将总RNA反转录为cDNA(反转录条件为25 ℃,10 min;42 ℃,50 min),1 μl的cDNA进行实时定量PCR反应。反应条件为变性94 ℃,10 s;退火60 ℃,20 s;延伸72 ℃,30 s;40个循环。72 ℃延伸150 s;40℃温育90 s;从60 ℃到94 ℃开始进行熔解,每秒1 ℃;25 ℃温育1~2 min。引物序列见表1。利用2-△△CT方法进行实时荧光定量分析。

RT-PCR引物序列
RT-PCR引物序列
| 基因 | 引物序列(5'-3') | 扩增片段长度(bp) |
|---|---|---|
| RET | R:TCAGAAGGTTGAAGAGCC | 106 |
| F:ACGCAAAGTGATGTATGGT | ||
| β-actin | R:TAGAAGCATTTGCGGTGG | 168 |
| F:GGCACCCAGCACAATGAA |
取感染48 h后细胞,全蛋白提取试剂盒提取蛋白,BCA法测定蛋白浓度,蛋白上样后进行SDS-PAGE电泳。使用PVDF膜,80 V恒压湿转,4 ℃一抗孵育过夜。TBST洗膜后37 ℃二抗孵育45 min后应用化学曝光法曝光显影。用凝胶图像处理系统(Gel-Pro-Analyzer软件)分析目标条带的吸光度值。
检测感染48 h后各组细胞中RET的定位情况。细胞爬片固定后0.1%tritonX-100透膜,1% BSA封闭,一抗4 ℃过夜孵育,荧光二抗室温孵育60 min。DAPI复染核。抗荧光淬灭剂封片,镜检。
检测感染24 h后各组细胞的迁移能力。制备细胞悬液稀释后显微镜下观察计数,计算后得到细胞悬液内含有的总细胞数。将已包被好的Transwell小室放入24孔板中,下室加入含10% FBS的培养液800 μl,分别取细胞悬液200 μl加入上室。将24孔板置于37 ℃、5%CO2、饱和湿度条件下的细胞培养箱中培养24 h。经固定并结晶紫染色,在倒置显微镜下对迁移至微孔膜下层的细胞计数,每个样本选取5个视野计数细胞个数,取均值。
检测感染5个时间点0、24、48、72、96 h后各细胞的增殖能力。每孔加入10 μl CCK-8,37 ℃,5%CO2的培养箱内培养2 h。在酶标仪上测定其在450 nm处的吸光度值(OD值),进行数据分析。
检测感染72 h后各组细胞的凋亡情况。收集细胞后制备悬浮细胞,加入AnnexinV-FITC,加入10 μl碘化丙锭,室温避光反应15 min,进行流式检测。
感染48 h后各组细胞用50 ng/μl重组人GDNF处理细胞15 min,Western blot检测各组细胞中p-Ret、p-ERK1/2和ERK1/2的蛋白表达。
所有符合正态分布的数据以 ±s来表示,应用Graph-Pad Prism 8.0.1统计软件进行统计学分析,多组均数间比较采用Tukey's T检验,多个研究组均数与对照组均数比较采用Dunnett-T检验,P<0.05表示差异有统计学意义。
±s来表示,应用Graph-Pad Prism 8.0.1统计软件进行统计学分析,多组均数间比较采用Tukey's T检验,多个研究组均数与对照组均数比较采用Dunnett-T检验,P<0.05表示差异有统计学意义。
RT-PCR检测显示各组RET mRNA的相对表达量,Control组为1.00±0.05,NC组为0.95±0.05,WT组为9.15±0.82,c.2599G>T组为7.25±0.35。与阴性对照相比,WT组表达升高,c.2599G>T组RET基因较WT组表达下降,差异有统计学意义(P<0.01)。Western blot实验结果如图1,在阴性对照组中未检测到RET蛋白表达,在WT组细胞可检测到全长RET蛋白,在c.2599G>T组细胞可检测到截短的RET蛋白。免疫荧光检测发现RET蛋白在野生过表达细胞和突变细胞中表达,表达于细胞膜及细胞质,突变细胞中荧光较野生过表达细胞略弱,如图2。
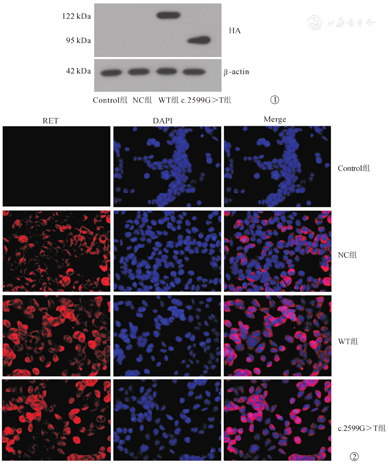
注:Control组,未感染组;NC组,阴性对照腺病毒对照组;WT组,RET野生型过表达腺病毒组;c.2599G>T组,RET c.2599G>T突变型过表达腺病毒组;在阴性对照组中未检测到RET蛋白表达,在WT组细胞可检测到全长RET蛋白,在c.2599G>T组细胞可检测到截短的RET蛋白;可见WT组和c.2599G>T组细胞在x细胞质和细胞膜有强烈的荧光信号,c.2599G>T组细胞中荧光信号较WT组略减弱
应用Transwell实验检测感染24 h后各组细胞的迁移能力。应用CCK-8实验检测感染5个时间点0、24、48、72、96 h后各细胞的增殖能力。应用流式细胞技术检测感染72 h后各组细胞的凋亡情况。各项结果见表2,图3。与阴性对照组相比,过表达野生型RET可以增强细胞的迁移和增殖能力,抑制细胞凋亡(P<0.001);与WT组相比,突变组的细胞迁移和增殖能力减弱,抑制细胞凋亡能力减弱(P<0.001)。
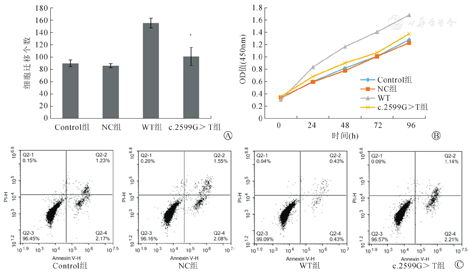
注:Control组,未感染组;NC组,阴性对照腺病毒对照组;WT组,RET野生型过表达腺病毒组;c.2599G>T组,RET c.2599G>T突变型过表达腺病毒组;a表示c.2599G>T组比WT组P<0.001;与WT相比,c.2599G>T组的细胞迁移和增殖能力减弱,抑制细胞凋亡能力减弱
各组细胞Transwell实验、CCK-8实验和流式细胞实验结果( ±s)
±s)
各组细胞Transwell实验、CCK-8实验和流式细胞实验结果(±s)
| 组别 | Transwell实验细胞个数 | CCK-8实验光密度值 | 流式细胞实验(%) | ||||
|---|---|---|---|---|---|---|---|
| 0 h | 24 h | 48 h | 72 h | 96 h | |||
| Control组 | 89.60±5.46 | 0.33±0.03 | 0.06±0.06 | 0.82±0.08 | 1.01±0.08 | 1.28±0.14 | 3.89±0.48 |
| NC组 | 85.60±3.36 | 0.34±0.04 | 0.60±0.06 | 0.78±0.08 | 1.01±0.13 | 1.23±0.10 | 4.01±0.41 |
| WT组 | 155.20±7.89 | 0.30±0.03 | 0.83±0.10 | 1.17±0.13 | 1.41±0.19 | 1.68±0.15 | 0.85±0.10 |
| c.2599G>T组 | 100.80±14.72 | 0.36±0.04 | 0.68±0.06 | 0.90±0.10 | 1.07±0.11 | 1.38±0.12 | 3.12±0.20 |
| P值 | <0.001 | 0.050 | 0.011 | 0.007 | 0.009 | 0.008 | <0.001 |
各组细胞经GDNF激活后,突变组细胞中p-Ret、p-ERK1/2的蛋白表达与野生型RET过表达组细胞中的表达呈明显下降(P<0.001)。Western blot结果如图4所示,灰度分析相对表达量数据如表3。各组间ERK1/2的蛋白表达差异无统计学意义。
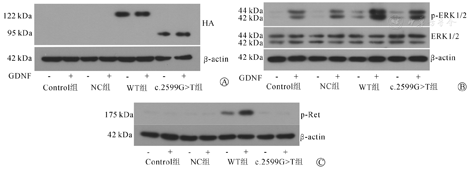
注:Control组,未感染组;NC组,阴性对照腺病毒对照组;WT组,RET野生型过表达腺病毒组;c.2599G>T组,RET c.2599G>T突变型过表达腺病毒组;各组细胞经GDNF激活后,突变组细胞中p-Ret、p-ERK1/2的蛋白表达与野生型RET过表达组细胞中的表达呈明显下降(P<0.001)
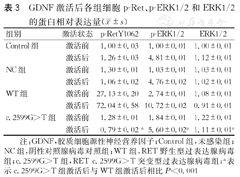
GDNF激活后各组细胞p-Ret、p-ERK1/2和ERK1/2的蛋白相对表达量( ±s)
±s)
GDNF激活后各组细胞p-Ret、p-ERK1/2和ERK1/2的蛋白相对表达量(±s)
| 组别 | 激活状态 | p-RetY1062 | p-ERK1/2 | ERK1/2 |
|---|---|---|---|---|
| Control组 | 激活前 | 1.00±0.03 | 1.00±0.01 | 1.00±0.01 |
| 激活后 | 1.26±0.03 | 4.81±0.01 | 1.12±0.01 | |
| NC组 | 激活前 | 1.30±0.01 | 1.03±0.01 | 1.03±0.01 |
| 激活后 | 1.06±0.02 | 4.76±0.02 | 1.02±0.01 | |
| WT组 | 激活前 | 27.13±0.20 | 2.74±0.01 | 1.08±0.01 |
| 激活后 | 72.04±0.58 | 10.72±0.02 | 0.91±0.01 | |
| c.2599G>T组 | 激活前 | 1.28±0.01 | 1.84±0.01 | 1.22±0.01 |
| 激活后 | 0.79±0.02 a | 5.60±0.02a | 1.11±0.01a |
注:GDNF,胶质细胞源性神经营养因子;Control组,未感染组;NC组,阴性对照腺病毒对照组;WT组,RET野生型过表达腺病毒组;c.2599G>T组,RET c.2599G>T突变型过表达腺病毒组;a表示c.2599G>T组激活后与WT组激活后相比P<0.001
HSCR是最常见的小儿消化道畸形,属于典型的肠神经系统(enteric nervous system,ENS)发育异常疾病。目前公认的发病机制是易感基因与环境因素共同作用,影响胚胎期肠神经元迁移,在细胞黏附因子等微环境的作用下,肠型中神经节细胞迁移、定植、发育和成熟发生障碍,导致HSCR的发生[8,9]。HSCR相关易感基因主要来自ENS发育过程中的信号通路[10,11]:①GDNF/GFRα1/RET信号系统;②RET基因转录调控的相关基因;③内皮素(endothelin,EDN)信号通路等。分析证实HSCR是一种遗传性疾病,可表现为常染色体显性(autosomal dominant,AD)、常染色体隐性和多基因形式。本例家系为AD,患儿母亲及2个患病女儿均为杂合突变。AD基因引起HSCR定位于染色体10q11.2,该区域有RET基因。
RET原癌基因有21个外显子,编码产物为传导信号的细胞表面分子,含有1 114个氨基酸的酪氨酸激酶受体蛋白。RET蛋白有3个结构域,细胞外配体结合结构域,跨膜结构域和细胞内酪氨酸激酶结构域。不同位置RET突变影响蛋白质功能的机制不同[12],细胞外结构域突变会使RET蛋白糖基化水平下降;细胞外半胱氨酸结构域突变会破坏RET蛋白糖基化,影响蛋白插入细胞膜;Y1062附近突变会破坏RET与其信号分子结合,阻碍RET信号通路的激活;增强子区域突变影响RET基因的表达量,导致细胞内RET基因mRNA水平降低。本研究组前期在1例巨结肠家系中检测到14号外显子无义突变c.2599G>T,14号外显子位于RET酪氨酸激酶结构域[7]。RET酪氨酸激酶结构域突变,将会影响RET蛋白质功能,会降低RET细胞内酪氨酸激酶活性,影响RET下游信号的转导。
目前RET基因突变占已有致病突变的80%以上,家族性HSCR中RET基因突变率可高达50%,散发病例在15%~35%。迄今为止,在HSCR患儿中发现的绝大多数RET突变均是基因测序技术获得的结果,仅有少数RET突变经过了体外功能检测。Widowati等[5]在检测到的9个RET变异中,只有3个显示出致病作用。所以,许多RET的突变没有进行功能测试,目前还很难辨别是否为有害或无害的突变。本课题组在前期结果基础上,在人胚肾细胞HEK-293中进行了过表达野生型RET基因和RET c.2599G>T突变细胞的功能学检测,证实了c.2599G>T突变是具有功能的有害突变。评估突变的功能意义非常重要,不仅可以有助于理解疾病的发病机制,而且可以提供准确的遗传咨询和复发风险评估。
HSCR是由于神经嵴细胞迁移失败引起的,神经嵴细胞迁移失败发生越早,无神经节长段越长。在迁移之前,有足够的细胞增殖才能使神经节细胞定居于远端肠管,如果迁移前的迷走神经嵴细胞减少,就会造成远端肠管的无神经节细胞改变,产生HSCR改变。RETc.2599G>T无义突变导致RET蛋白质截短,截短后影响了细胞增殖、迁移和抑制凋亡的功能,可能造成神经细胞增殖和迁移能力下降,凋亡增加,造成肠神经系统发育异常,远端肠管无神经节细胞,形成HSCR的病理改变。应用GDNF激活细胞,p-Ret、p-ERK1/2的蛋白相对表达明显下降,证实发生在RET酪氨酸激酶区的c.2599G>T突变可以导致RET酪氨酸激酶活性丧失,从而使得激动信号无法传输,进一步损害RET磷酸化过程,导致下游ERK的磷酸化被抑制,使得神经节细胞凋亡,诱发HSCR的形成。总之,肠道神经节细胞的发育、分化、移行和定位是靠RET接受激动信号来维持的,如果RET发生功能性突变,必然会使这一信号传输过程受到影响,从而干扰神经节细胞的分化过程,引起HSCR的发生。
本例家系母亲和2例女患儿虽然都有RET c.2599G>T的杂合突变,但是3人的表型轻重有所不同,母亲表现为便秘,大女儿表现为常见型巨结肠,小女儿表现为全结肠型巨结肠。目前研究认为环境因素、表观遗传或随机因素导致了不完全外显率和可变表达,分析可能存在以下几点原因:①RET基因启动子区富含5'-CG-3'序列,其表达受DNA甲基化的调控[13];②RET基因与其他基因相互作用,Nrg1、PHOX2B和SOX10等很多基因可通过对复杂的网络作用干扰RET信号[10,11,14];③miRNA和lncRNA等非编码RNA通过与靶基因互补结合触发其mRNA的降解或抑制其翻译来调控RET的表达,导致HSCR的发生[15,16];④基因表达调控是生物个体发育的重要基础,基因在生物个体发育过程中应该作为基因调控网络的一部分来看待,胚胎发育期人类肠道中可能有其他转录因子或增强子在RET基因的调控网络中发挥作用[17,18,19]。总之,HSCR是典型的多个基因共同参与,同时与环境因素相关的复杂人类遗传病,该家系中临床表型轻重不一的原因尚需进一步研究。本研究的局限性在于:①在多种细胞系中研究RET基因c.2599G>T的功能将会更有意义;②尚未构建RET基因c.2599G>T敲除动物模型;③亟待扩大样本量,在散发的HSCR病例和正常人群中检测RET基因c.2599G>T的发生频率。
随着基因检测技术和相关功能研究手段的发展,越来越多的突变在HSCR患儿家系中被发现和鉴定,当突变数据用于重要的临床决策如产前诊断时,功能分析是必要的。功能研究也将使人们对HSCR的发病机制有更深入的了解,对HSCR的临床治疗、遗传咨询及肠神经系统发育的研究有重大意义。
所有作者均声明不存在利益冲突

























