
2022年WHO在线(https://tumourclassification.iarc.who.int/home/)发布了第五版儿童肿瘤分类,外周神经母细胞性肿瘤(peripheral neuroblastic tumours,pNTs)作为独立章节被介绍。本次儿童肿瘤分类新增免疫组织化学对节细胞神经瘤与节细胞神经母细胞瘤的鉴别作用,细化pNTs的形态特征,强调形态学对遗传变异以及预后的提示意义、MYC家族蛋白表达较基因扩增更强的指示价值等。本文结合相关文献,对第五版WHO儿童肿瘤分类pNTs做简要介绍,以期为病理医生和临床医生提供新的诊疗帮助。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
第五版世界卫生组织(World Health Organization,WHO)儿童肿瘤分类在线预印版于2022年发布(https://tumourclassification.iarc.who.int/home/),该版儿童肿瘤分册共15个章节分别介绍儿童各系统肿瘤特征,其中外周神经母细胞性肿瘤(peripheral neuroblastic tumours,pNTs)作为独立章节被重点介绍。pNTs曾经归属于WHO神经系统肿瘤分类,第四版WHO肿瘤分类将其归入内分泌肿瘤分册中,即将发行的第五版WHO内分泌和神经内分泌肿瘤分类(以下简称"第五版内分泌肿瘤分类")仍继续保留。第五版WHO儿童肿瘤分类对pNTs病因学、临床表现、组织学、遗传学以及预后等方面做了详细描述,新增免疫组织化学(immunohistochemistry,IHC)对节细胞神经瘤与节细胞神经母细胞瘤的鉴别作用,细化pNTs的形态特征,强调形态学对遗传变异以及预后的提示意义、MYC家族蛋白表达较基因扩增更强的指示价值等。现就2022年第五版WHO儿童肿瘤分类pNTs做简要介绍。
pNTs代表一组由不同分化程度的神经母细胞和施万细胞构成的肿瘤。在第四版和第五版内分泌肿瘤分类中,该章节名称为肾上腺神经母细胞性肿瘤。第五版WHO儿童肿瘤分类使用了其广义的命名即pNTs,其分为四个亚型:神经母细胞瘤(neuroblastoma,NB);节细胞神经母细胞瘤,混杂型(ganglioneuroblastoma,intermixed,GNBi);节细胞神经母细胞瘤,结节型(和其他复合性神经母细胞性肿瘤)(ganglioneuroblastoma,nodular,GNBn);节细胞神经瘤(ganglioneuroma,GN)。第五版WHO儿童肿瘤分类遵从第五版内分泌肿瘤分类统一版式,对每种亚型分别从定义、临床特征、病因学、组织病理学及预后等方面进行详细介绍。
第五版WHO儿童肿瘤分类和第五版内分泌肿瘤分类介绍pNTs起源于原始神经嵴细胞,原始神经嵴细胞在正常情况下发育为肾上腺髓质和交感神经节,这也是第五版内分泌肿瘤分类将pNTs与副神经节瘤、嗜铬细胞瘤归于同一大类的原因。儿童肿瘤分类进一步强调,pNTs是由这种调节正常交感神经系统发育的分子机制缺陷引起。肾上腺髓质或肾上腺外副神经节中的交感神经元和嗜铬细胞可能来自不同的前体细胞,交感神经节通过其前体从神经嵴直接迁移而来,而嗜铬细胞主要来自被称为"施万细胞前体"(Schwann cell precursors,SCPs)的神经嵴细胞,在神经嵴消失后,SCPs可作为神经嵴细胞的储存库。Kastriti等[1]的研究发现,NB可能重新激活了发育相关的基因表达,从而表现为SCPs样特征。国内Dong等[2]通过单细胞转录组测序证明,大多数肾上腺NB可能来源于去甲肾上腺素能嗜铬细胞,其恶性状态类似于嗜铬细胞在正常发育过程中的增殖和(或)分化状态。这些新的发现为阐明pNTs的分布、发病年龄以及肿瘤成熟等提供依据。
NB是pNTs中最多见的一类,为1岁以内儿童最常见肿瘤。原发部位沿神经嵴发育迁移路径分布,包括颈部、胸部、腹部、肾上腺和盆腔的交感神经节。NB可罕见发生于睾丸旁[3],甚至可无原发病灶[4]。NB临床表现存在异质性,肿瘤位于腹部者可引起腹胀和(或)便秘,位于纵隔者可引起呼吸窘迫,当脊柱旁肿瘤通过椎管侵入硬膜外可引起神经症状,转移瘤可引起淋巴结病、肝肿大、骨痛、"熊猫眼"等。罕见情况下,具有PHOX2B突变的患儿表现为先天性中枢性通气不足和先天性巨结肠[5]。
目前普遍认为NB是由遗传或表观遗传事件驱动,比较公认的有MYC家族癌基因过表达、ALK基因过表达和异常端粒维持和(或)延长。然而临床上对NB的确切病因仍知之甚少。低龄发病和原位NB提示肿瘤始于胎儿期,其中原位NB是在婴儿尸检肾上腺中发现,仅显微镜下可见,NB灶不伴有转移[6]。研究发现,TH-MYCN小鼠模型在妊娠期接受应激刺激,其子代发生NB的概率增加,且肿瘤出现肺转移,致死率亦明显升高,提示母体应激可能是促进NB进展的产前因素[7]。研究人员通过对556例NB患儿和其父母的配对全基因组测序发现,13.5%(75/556)的NB存在遗传性致病或可能致病的胚系突变,除常在家族性NB中出现的ALK和PHOX2B,被鉴定出的肿瘤易感基因还包括BARD1,这种父母携带同样位点的变异而不发生NB的遗传学机制有待进一步揭示[8]。关于驱动肿瘤的分子遗传因素,第四版内分泌肿瘤分类已介绍,而第五版WHO儿童肿瘤分类与第五版内分泌肿瘤分类再次强调:虽然大部分肿瘤过表达MYCN蛋白,但存在少部分过表达MYC(又名C-myc)蛋白,这类肿瘤很少出现MYCN基因扩增[9]。并且第五版WHO儿童肿瘤分类进一步阐释了这种非基因扩增所致MYC过表达的其他分子机制。研究显示其可能通过易位劫持增强子激活癌基因,也可通过MYCC编码区(8q24)附近的焦点增强子扩增或MK2介导的OCT4转录激活MYCC基因[10,11]。过表达MYC家族蛋白(MYCN或MYC)的肿瘤被统称为MYC驱动的NB[12]。
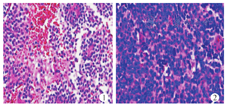
NB由薄纤维血管间隔形成巢状结构,根据瘤巢内神经母细胞的分化程度不同,可分为未分化、差分化和分化型。未分化型因肿瘤细胞呈均匀一致的"小圆蓝细胞"形态,缺乏可辨识的神经毡结构,常需IHC和(或)分子检测确诊。差分化型背景可见神经毡结构,可有Homer Wright菊形团结构形成,少于5%的神经母细胞呈分化的形态特征,肿瘤细胞核呈"椒盐"样外观(图1);也可表现为少见的细胞形态,如大而多形、梭形以及假横纹肌样细胞。分化型肿瘤具有丰富的细胞毡,超过5%的细胞呈分化形态,即细胞核(增大、偏位,泡状染色质以及单个大核仁)和细胞质(嗜酸或嗜双色性,直径大于2倍细胞核)同步分化。同时,第五版WHO儿童肿瘤分类与第五版内分泌肿瘤分类保持一致,再次细化形态学特征可反映基因改变的情况。MYCN扩增的肿瘤在组织学上常呈未分化或差分化型,有丝分裂-核碎裂指数(mitosis-karryorrhexis index,MKI)高。MYC驱动的肿瘤常常有明显核仁,可能是由MYCN或MYC的RNA合成和积聚所致[13],形态上可与其他含"椒盐"样核的肿瘤区别。还有一类罕见的大细胞NB,细胞核大、染色质开放,包含一至多个明显的核仁(图2),表明MYCN或MYC蛋白的高表达水平,这种富含常染色质的细胞核也提示肿瘤的干性特征[14]。大细胞NB的细胞核形态与分化的神经母细胞核类似,二者应严格区分,大细胞NB细胞核比常规NB细胞核大、细胞质稀少,而分化的神经母细胞则具有2倍于细胞核的丰富细胞质[15]。另外,此前被归为大细胞NB的大核仁神经母细胞瘤(large nucleolar neuroblastoma,LNN),以大而显著的核仁和极其稀少的细胞质为特征,也是MYCN扩增的组织学表征[15]。
免疫学表型上,第五版WHO儿童肿瘤分类与第四版、第五版内分泌肿瘤分类均提到除常规神经源性标志物外,PHOX2B有重要的诊断应用价值。PHOX2B对NB(尤其是未分化型)具有高特异性和敏感性,可有效鉴别NB与其他具有"小圆蓝细胞"形态的肿瘤[16]。且PHOX2B在治疗后的病灶以及骨髓转移灶中亦呈明显的核表达,识别作用显著[17]。NB被分为组织学良好型(favorable histology,FH)和组织学不良型(unfavorable histology,UH),UH又被分为四个亚组即MYC组、TERT组、ALT组和Null组;免疫标志物MYCN/MYC、TERT、ATRX表达水平可有效识别NB分组,有助于确定精准治疗的相应靶点[18]。其中,ATRX的IHC染色不能完全识别由ATRX突变所致的ALT表型,可辅以荧光原位杂交(fluorescence in situ hybridization,FISH)帮助识别,尤其是对5岁以上的ALT表型患儿,可从抗ALT治疗中获益[19]。罕见情况下,ALT表型伴由基因突变所致的DAXX缺失[20]。因此,第五版WHO儿童肿瘤分类推荐有条件时应行基因检测,包括MYCN以及ALK癌基因状态、端粒维持基因(TERT和ATRX)状态、DNA倍性和节段性染色体畸变。
对于NB的预后预测主要依据临床分期、诊断年龄和组织学。第五版WHO儿童肿瘤分类强调特殊组织学对预后具有一定的预测作用。如Tornóczky等[21]的研究中7例患儿组织学上为特殊形态的大细胞NB,具有高度侵袭性,4年内患儿全部死亡。同样地,LNN亦具有高侵袭性,患儿预后极差[15]。究其原因,可能是由于这类肿瘤由MYC驱动所致。研究认为,MYC蛋白过表达可能比基因扩增更能预示肿瘤的强侵袭性[22]。此外,分子遗传学特征对个体肿瘤的预后预测意义重大,需注意的是,DNA倍性预测不适用于超过2岁的患儿,因为此时肿瘤通常伴有染色体结构性异常;各种节段性染色体畸变中6q缺失见于侵袭性极强的肿瘤[23,24]。对于ALK基因异常,第四版内分泌肿瘤分类认为可致NB预后差,Emily等[8]的研究支持该说法。作为两大主要致病或可能致病的肿瘤易感基因,ALK和PHOX2B胚系变异与NB患儿预后更差相关。但第五版WHO儿童肿瘤分类对此观点提出质疑。例如携带ALK胚系突变的同一家族患儿,预后结局大不相同,并不总是差的[25];还有ALK突变的NB发生自发消退的报道[26];超深度测序揭示激活性ALK突变与患儿生存率无明显相关[27]。
GNBi是一种介于NB和GN之间的过渡形式,由施万细胞和散在其中的NB灶构成,NB灶内神经母细胞可处于不同成熟阶段。与NB的低龄发病不同,GNBi好发于平均年龄4~7岁的儿童。临床上常表现为无症状包块。第五版WHO儿童肿瘤分类突出了影像学的作用,GNBi在横断面成像上显示为包裹性边界清楚的肿块,弥散加权磁共振成像显示GNBi的表面弥散系数明显高于NB,有助于二者鉴别[28]。此外,与NB呈间位碘代苄胍高摄取(90%)不同,GNBi的间位碘代苄胍摄取率低至34%[29]。
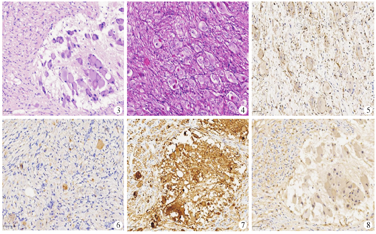
作为对于GNBi的重要更新,第五版WHO儿童肿瘤分类指出IHC标记S100和Syn对GNBi和GN具有鉴别诊断意义。除含有神经毡结构的NB灶外(图3),GNBi中可出现类似GN的形态,表现为成簇的节细胞位于S100阳性、Syn阴性的施万细胞基质中(图4,图5,图6)。而GNBi内的神经毡呈Syn阳性(图7)、S100阴性(图8),IHC检测有助于识别神经毡结构,从而与GN鉴别。预后方面,GNBi通常为区域局限性肿瘤,被划分为低风险,极少转移扩散,且不具有化疗敏感性。值得注意的是,仍有部分为转移性病例,这类GNBi预后较差,存在一定的复发和死亡风险。
GNBn是在GN背景上出现一个或多个肉眼可见神经母细胞结节。GNBn发病年龄亦较NB大,多大于18个月。临床表现通常为局限的腹部包块,伴腹痛、腹胀和发热,也可呈广泛转移。一般而言,GNBn组织呈黄褐色、质坚实,常含单个出血性或坏死性结节,少数可为多个结节,边界清楚。第四版内分泌肿瘤分类仅少量篇幅介绍了GNBn的常规组织学表现,第五版WHO儿童肿瘤分类对其做了更为详细的描述。原发肿瘤无肉眼可见的NB结节,仅转移灶表现为GNBn是十分罕见的[30]。神经母细胞结节可能起源于后天获得的遗传变异引起的克隆性增生[31,32]。也有观点认为神经母细胞结节与周围GN在遗传学上具有相似性,并非来源于不同的克隆[33]。
GNBn的组织学诊断有赖于识别结节性神经母细胞成分和非结节以施万细胞为主的成分。结节包括未分化、差分化或偶尔分化的神经母细胞,神经母细胞结节周围可为GN或GNBi,当结节过度生长,少量的GN成分可仅残存于肿瘤周边区。最常用的pNTs预后分类标准为国际神经母细胞瘤病理分类(international neuroblastoma pathology classification,INPC),第五版WHO儿童肿瘤分类强调GNBn的INPC分类应依据结节性神经母细胞成分,因为患儿的预后主要取决于这部分肿瘤细胞的侵袭情况。当有多个结节存在时,每个结节都需单独评估。因GNBn的异质性,单独部位的针穿活检很容易遗漏GNBn中的其他结节,导致误诊,延误治疗。因此,全面取材在活检和大体检查中均是必不可少的。对于其他复合性神经母细胞性肿瘤,第五版WHO儿童肿瘤分类解释其表现为多个不同的神经母细胞成分,这种多克隆性在组织学、IHC和遗传学方面均有体现,有待进一步确定其预后因素。
GN代表了pNTs谱系中分化成熟的一类,可能由早期的NB发展而来。与NB好发于肾上腺不同,GN常发生于胸部,临床上多无明显症状,常偶然发现。GN发病年龄与GNBi相近。第五版WHO儿童肿瘤分类认为GN的产生机制可能是由于生物学良好的神经母细胞产生了刺激施万细胞增殖和发育的关键因子,趋化其向肿瘤内迁移,这些进入肿瘤的施万细胞反过来分泌抗增殖和诱导分化的因子,从而促进神经元成熟[34,35]。
GN组织形态表现为丰富温和的施万细胞(>50%肿瘤体积)排列成片状或束状,分散或成簇分布正在成熟和(或)成熟的节细胞。正在成熟的节细胞大小和形态各异,可存在多个细胞核。第五版WHO儿童肿瘤分类介绍GN中散在分布正在分化的神经母细胞,但不成巢且不含神经毡结构是非常少见的,因此有别于NB和GNBi。其他组织学表现还有成熟的节细胞常被卫星细胞围绕,淋巴细胞浸润也是比较常见的现象。GN通常瘤体较局限、临床分期低,即便是在不完全切除的情况下,预后也是极好的。
综上所述,2022年WHO儿童肿瘤分类中pNTs新增IHC对GN与GNBi的鉴别作用,细化pNTs的形态特征,强调形态学对遗传变异以及预后的提示意义、MYC家族蛋白表达较基因扩增更强的指示价值等。其中GNBi与GN形态上可部分重叠,因GNBi不完全良好的预后特征,临床工作中需与GN进行鉴别,IHC标志物S100和Syn有助于二者区分。特殊的NB组织学可呈现大核、开放的染色质以及显著的核仁,往往提示MYCN或MYC蛋白的高表达以及MYCN基因扩增,同样也预示着肿瘤的高侵袭性和极差的预后。MYC蛋白与MYCN蛋白均具有重要的NB驱动作用,该MYC家族蛋白过表达比基因扩增更能预测肿瘤预后。
所有作者均声明不存在利益冲突

























