
总结Duchenne肌营养不良症新生儿临床表型和基因突变特点。
回顾性分析福建医科大学附属福州市第一医院新生儿科收治的1例DMD基因新发缺失突变引起的Duchenne肌营养不良症新生儿临床表型和基因突变特点。
患儿临床表型为出生后低血糖、低体温、反应欠佳、肌张力尚可,出生半月内天冬氨酸转移酶、肌酸激酶、肌酸激酶同工酶和乳酸脱氢酶等各类心肌酶检测均较正常值高,心电图显示窦性心律和部分导联T波改变。患儿母亲、父亲和哥哥出生时均正常、未见患儿样临床表现,目前该家系除患儿外均健康。基因检测显示:患儿DMD基因47号外显子存在一个遗传自母亲的杂合变异c.6863_6870delinsTTG(p.Gln2288Leufs*3),为缺失突变,患儿父亲和哥哥未携带该变异,经检索为新发突变,结合患儿临床症状确诊为Duchenne肌营养不良症。
DMD基因外显子47 c.6863_6870delinsTTG(p.Gln2288Leufs*3)为新发缺失突变,丰富了DMD基因突变谱,为Duchenne肌营养不良症遗传咨询和产前诊断提供了有价值的信息。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Duchenne肌营养不良症(Duchenne muscular dystrophy,DMD),又叫假肥大型肌营养不良症,是一种常见的X-连锁隐性遗传、致死性肌肉疾病,由位于Xp21的抗肌萎缩蛋白基因突变而引起抗肌萎缩蛋白完全或部分缺失所致[1]。流行病学统计数据显示,全球平均每3 500个新生男婴中就有1人罹患DMD,女性多为致病基因携带者,发病者罕见,且症状较轻;我国每年约有400~500例DMD患儿出生,累计约7万人确诊为DMD,是世界上该病患者人数最多的国家之一[2,3]。DMD患儿在学龄前就会因骨骼肌不断退化出现肌肉无力或萎缩,导致行走不便,大概在7~12岁时,会彻底丧失行走能力,通常到20多岁就会因为心肌、呼吸肌无力而死亡[1]。遗憾的是,到目前为止医学界针对DMD尚无有效的治疗方法,所以对高风险人群家系的遗传咨询和产前基因检测将有助于减少DMD患儿的出生和降低疾病发作对患儿及其家庭造成的伤害[4]。本研究通过对福建医科大学附属福州市第一医院新生儿科收治的1例疑似DMD的新生儿及其母亲、父亲、哥哥进行全外显子组测序,分析其临床表型和基因突变特点,现报道如下。
患儿,男,2 d,主诉"发现血糖低5 h,体温不升1 h"入院。患儿为G2P2,胎龄38+6周,在本院产科顺产出生。出生体重2.89 kg。出生后快速评估,足月,前羊水清,后羊水血性,呼吸尚可,四肢发绀,肌张力尚可,立即予保暖、擦干、刺激等处理,Apgar评分:1 min评9分(肤色扣1分),继续予保暖等处理,5 min、10 min均评10分。出生后,每隔1小时检测患儿末梢血糖分别为2.0 mmol/L、1.3 mmol/L和2.5 mmol/L,嘱继续观察。出生5 h,患儿出现反应欠佳,嗜睡,面色及口唇稍发绀,四肢冰凉,甲床紫绀,哭声弱,吸吮欠佳,测体温35.2 ℃,予保暖、复温等处理后,复测体温35.3 ℃,测末梢血糖3.3 mmol/L;患儿无呻吟、气促,无呕吐、腹胀,无惊厥、意识不清。患儿无其他病史,否认母孕期疾病、否认双亲近亲结婚,否认家族其他成员类似病史。
入院查体:体温35 ℃;脉搏114次/min;血压66/36 mmHg(1 mmHg=0.133 kPa);血氧饱和度:动脉导管前93%,动脉导管后92%。反应欠佳,嗜睡,少哭。背部可见散在少许红色针尖样出血点,压之不褪色,其余皮肤黏膜无黄染、皮疹。前囟平软。双侧眼睑无水肿,双侧结膜无充血,巩膜无黄染,双侧瞳孔等大等圆,直径约为2.5 mm,对光反射灵敏。双耳廓无畸形。鼻翼无扇动。面色及口唇稍发绀。颈部无抵抗感,气管居中。胸廓对称,三凹征阴性,双肺呼吸音粗,未闻及干湿性啰音。心前区无病理性隆起、无震颤,无心包摩擦感,心律齐,心音低钝,心前区听诊区可闻及Ⅰ/Ⅳ级收缩期杂音。腹部稍膨隆,无胃型、肠型及蠕动波,无腹壁静脉曲张,脐部已结扎,腹软,未触及肿物,肝右肋下1 cm,肠鸣音4次/min。毛细血管充盈时间3~4 s,甲床紫绀,肢端冰凉。四肢肌张力尚可,吸吮、吞咽反射减弱,觅食及拥抱反射存在。
辅助检查及治疗过程:患儿出生当天中性粒细胞百分比(63.5%)升高,凝血酶原时间(15.0 s)、活化部分凝血活酶时间(48.9 s)升高,凝血酶原活动度(73.1%)、纤维蛋白原(1.02 g/L)降低,D-二聚体(1.15 mg/L)、纤维蛋白原降解产物(5.30 μg/mL)、乳酸(3.80 mmol/L)、白细胞介素-6(21.2 pg/mL)、谷草转氨酶(255 U/L)、肌酸激酶(38 500 U/L)、肌酸激酶同工酶(184.61 ng/mL)、乳酸脱氢酶(1 668 U/L)、肌酐(76 μmol/L)、葡萄糖(2.62 mmol/L)、总胆红素(60.17 μmol/L)、直接胆红素(6.62 μmol/L)、间接胆红素(53.55 μmol/L)升高。此外,心电图检测提示窦性心律和部分导联T波改变(图1);脑电图检测显示双侧脑区可见少许尖波发放,轻度异常振幅整合脑电图。床旁超声检查:动脉导管未闭7.0 mm×2.2 mm,卵圆孔未闭1~2 mm和三尖瓣轻度反流。
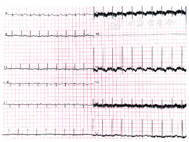
窦性心律和部分导联T波改变。
患儿入院后予心电监护、辐射台复温、头孢噻肟钠预防感染、1,6-二磷酸果糖营养心肌、凝血酶原复合物改善凝血、多巴胺及多巴酚丁胺[6 μg/(kg·min)]改善循环、氨溴索促进肺成熟、维生素C改善心肌细胞代谢、补液等处理。经治疗后,各项实验室指标均恢复正常,但患儿出生后第1、4、8、15天天冬氨酸转移酶、肌酸激酶、肌酸激酶同工酶和乳酸脱氢酶等各类心肌酶检测均较正常值高。
对症治疗1个月后,取患儿及患儿父亲、母亲和哥哥外周血进行全外显子组测序,结果显示:患儿DMD基因47号外显子存在一个遗传自母亲的杂合突变c.6863_6870delinsTTG(p.Gln2288Leufs*3),为缺失突变,患儿父亲和哥哥未携带该变异。该变异在正常参考人群基因数据库未见报道;计算机辅助分析预测该变异影响蛋白质结构/功能可能性较大(图2)。
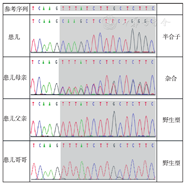
随访:患儿出生后11个月入院进行随访,谷丙转氨酶(279 U/L)、谷草转氨酶(397 U/L)、肌酸激酶(17 737 U/L)、肌酸激酶同工酶(271.46 ng/mL)、乳酸脱氢酶(1 483 U/L)、促甲状腺素(13 410 mIU/L)升高;经《0~6岁小儿精神发育诊断量表》《婴幼儿智能发育量表》《盖瑟尔智能发育诊断量表》评估,精神智能发育正常;经《婴儿-初中生社会生活能力量表》评估社会适应能力正常。
目前,DMD的致病机制已经十分明确,即由DMD基因突变引起。DMD基因编码dystrophin蛋白,dystrophin主要分布于骨骼肌和心肌细胞,可以保护肌细胞膜在肌肉收缩时不受损伤[5]。DMD基因突变会引起多种肌肉系统疾病,根据突变的位点不同,患病的严重程度有所差别,DMD为其中较为严重的一种病症[6,7]。
本例患儿出生后表现为低血糖、体温不升、反应欠佳、心电图轻微异常、心肌酶谱升高、总胆红素和间接胆红素升高,并且超声提示该患儿动脉导管未闭、卵圆孔未闭以及三尖瓣轻度反流,符合DMD临床特征;但该患儿母亲、父亲和哥哥均健康。经对患儿及其父母、哥哥的外周血DNA进行全外显子组测序发现:患儿DMD基因47号外显子存在一个遗传自母亲的杂合变异c.6863_6870delinsTTG(p.Gln2288Leufs*3),为缺失突变,可以解释患儿表型的疑似致病性变异。研究表明,男性患儿的DMD基因致病变异多来自母亲,女性DMD基因致病突变携带者的后代中,男性有50%可能患病,50%可能正常;女性50%可能为携带者,50%可能不携带致病突变[8,9,10]。因此,对女性DMD基因携带者进行产前基因检测和遗传咨询可有效降低DMD患儿的出生率。
DMD基因是迄今为止发现的最大人类基因,编码dystrophin蛋白,该蛋白有79个外显子,共有3 685个氨基酸[11]。DMD基因致病性变异会导致抗肌营养不良蛋白的缺乏或减少,肌纤维的完整性遭到破坏,进而引发肌纤维坏死、肌肉纤维化和再生能力丧失,严重者可见纤维结缔组织和脂肪组织替代正常的肌肉组织,出现肌肉假性肥大的典型表现[12,13]。已经报道的DMD基因变异类型包括外显子缺失、重复及点突变等,其中大片段缺失/重复约占60%~70%,其余为点突变或微小缺失/重复[14,15]。本研究患儿为DMD基因外显子47 c.6863_6870delinsTTG(p.Gln 2288Leufs*3)缺失突变,为新发突变,在千人基因组数据库、ESP6500数据库、Exome Aggregation Consortium(ExAC)数据库和ClinVar数据库中均不存在这种缺失突变。文献报道,DMD基因以44~47外显子缺失突变最为常见[16,17]。本研究患儿DMD基因47号外显子缺失7个核苷酸的情况是首次被报道,经生物信息学分析该突变可能激活无义介导的mRNA降解,从而影响该基因编码蛋白质的功能。
目前,治疗DMD患者的方法主要包括药物治疗、基因治疗和康复治疗等,药物治疗临床上广泛采用皮质激素,但其不良反应较大。基因治疗是目前最有希望治愈DMD的疗法之一,主要包括外显子跳跃、基因递送、基因编辑以及终止密码子通读等几种类型。此外,运动康复治疗和物理康复治疗也能有效延长DMD患儿的生存时间。然而,目前的治疗方法均无法治愈患者,产前遗传咨询和基因检测是目前预防DMD的最有效方案[4]。
所有作者均声明不存在利益冲突

























