
分析MYH9相关疾病的临床表型及基因突变特征。
回顾性分析2010年1月至2022年12月苏州大学附属第一医院66例MYH9相关疾病患者临床资料。根据有无出血将患者分为出血组和未出血组,根据突变位点将患者分为非肌性肌球蛋白重链ⅡA头部区域(MD)突变组和尾部区域(TD)突变组。统计分析不同组别患者血小板数量、出血情况、肾功能、白内障及听力等基本临床表型特征以及MYH9基因突变情况。
66例MYH9相关疾病患者中,其中男28例,女38例,确诊年龄为1~63(26±2)岁,41%(27/66)为散发病例。所有患者均出现血小板减少伴有巨大血小板,血小板聚集功能(10/10)均正常。92%(54/59)患者中性粒细胞内可见蓝色的包涵体,30%(20/66)患者存在出血,45%(22/49)患者存在蛋白尿或肾小球肾病,20%(8/41)患者存在双侧听力受损,10%(4/42)患者存在双侧白内障。共发现18个基因突变位点,包括错义突变15个、剪切突变1个、终止突变2个,其中56%(29/52)患者存在p.Asp1424Asn、p.Arg1933*及p.Arg702His/Cys突变,p.Ser96Leu、Arg1165Cys及p.Glu1841Lys突变为重现型突变,p.Ala44Thr、p.Asp1447Ala及c.3838-2A>G突变为新发突变。出血组患者血小板数量为(19±3)×109/L,明显少于未出血组的(36±3)×109/L(P<0.001)。与TD突变组比较,MD突变组血小板数量更少,出血等级更高,肾脏受累程度、听力受损及白内障等临床表型症状更重(均P<0.05)。
p.Asp1424Asn、p.Arg1933*及p.Arg702His/Cys为MYH9相关疾病患者的热点突变。MD突变组血小板数量更少,出血等级更高,肾脏受累程度、听力受损及白内障等临床表型症状重于TD突变组。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
MYH9相关疾病(MYH9-related disorder,MYH9-RD)是一类以血小板减少、巨大血小板及中性粒细胞包涵体为特征的常染色体显性遗传性疾病[1, 2]。多数患者以血小板减少就诊,部分患者表现为自发性出血,另外尚有部分患者伴发蛋白尿或肾小球肾病、听力受损以及白内障[3, 4]。由于患者的临床表现具有显著异质性,早期根据不同的临床表现组合将该病分为4种不同疾病:May-Hegglin异常、Sebastian综合征、Fechtner综合征和Epstein综合征[5]。随着测序技术的应用,明确以上4种疾病均是由MYH9基因突变所致,是一种疾病的不同亚型,因此2004年MYH9-RD的命名被广泛应用[6, 7]。MYH9-RD发病率约为3/10万,约35%为散发病例,是遗传性血小板减少症中最常见的类型。由于临床认识不足,许多患者被误诊为慢性免疫性血小板减少并接受免疫抑制及切脾等不当治疗[8]。国内目前已有多篇MYH9-RD报道,但病例有限,本研究旨在总结分析MYH9-RD患者临床表型及基因突变特征,以提高对该病的认识。
本研究为病例系列研究,经苏州大学附属第一医院伦理委员会审核同意并批准(批号:2023伦研批第279号),且豁免患者知情同意。收集2010年1月至2022年12月苏州大学附属第一医院血液科MYH9-RD患者66例。纳入标准:(1)血小板减少伴有巨大血小板(<100×109/L),血涂片示中性粒细胞异常包涵体;(2)血小板减少且基因检测显示存在已知MYH9基因突变;(3)大血小板(直径>3.9 μm的血小板占40%以上)减少、未见中性粒细胞包涵体,且存在未知MYH9基因突变;(4)确诊MYH9家系伴有血小板减少的成员。根据有无出血将患者分为出血组和未出血组;根据突变位点将患者分为非肌性肌球蛋白重链ⅡA头部区域(MD)突变组和尾部区域(TD)突变组。
1.表型分析:收集MYH9-RD患者的临床资料,包括性别、年龄、出血、肾功能、白内障及听力情况等。(1)血小板数量分级评估:1级:血小板数量>50×109/L;2级:20×109/L<血小板数量≤50×109/L;3级:血小板数量≤20×109/L。(2)出血等级评估:根据WHO出血程度分级标准(修改版)评价自发性出血严重程度。(3)肾脏受累程度评估:0级:尿蛋白正常;1级:尿蛋白+;2级:尿蛋白++;3级:尿蛋白+++;4级:尿蛋白++++、需透析或已行肾脏移植。
2.外周血细胞分析:采用血细胞分析仪检查血小板数量;采集患者末梢指端血制备血涂片,经瑞氏染色后光学显微镜观察血小板数量、大小以及中性粒细胞有无包涵体。
3.血小板聚集功能分析:取富血小板血浆(platelet-rich plasma,PRP),在诱导剂二磷酸腺苷(adenosine diphosphate,ADP)及瑞斯托霉素作用下,采用血小板聚集仪检测血小板最大聚集率。
4.MYH9基因测序分析:提取基因组DNA,采用出血血栓疾病靶向基因测序组套对先证者和其家属进行基因检测。将提取的DNA用超声波打断并制备DNA文库,通过捕获芯片对靶向基因的外显子及其临近的内含子区域的DNA进行富集,采用通用引物对捕获的序列进行PCR扩增,在IonS5测序平台(美国赛默飞公司)对扩增产物进行二代测序(next-generation sequencing,NGS)检测。最后使用IGV软件可视化分析NGS数据中的基因组变异情况。
采用SPSS 25.0软件进行统计分析。正态分布的计量资料以表示,组间比较使用两独立样本t检验。计数资料以频数及百分比表示,组间等级资料的比较采用Mann-Whitney U检验。出血和血小板数量的相关性采用Spearman相关性分析。双侧检验,检验水准α=0.05。
66例MYH9-RD患者来自45个不相关家系,27例(41%)为散发病例。其中男28例,女38例,确诊年龄为1~63(26±2)岁。所有MYH9-RD患者均存在血小板减少,30%(20/66)的患者存在出血。10例患者进行了血小板聚集功能测定,其ADP及瑞斯托霉素诱导的血小板聚集功能均正常。59例患者进行了外周血涂片分析,其中中性粒细胞内可见蓝色包涵体的占92%(54/59)。45%(22/49)患者存在肾脏受累,其中2例患者死于肾功能衰竭,2例进行肾脏移植手术。20%(8/41)患者存在双侧听力受损,10%(4/42)患者存在双侧白内障。
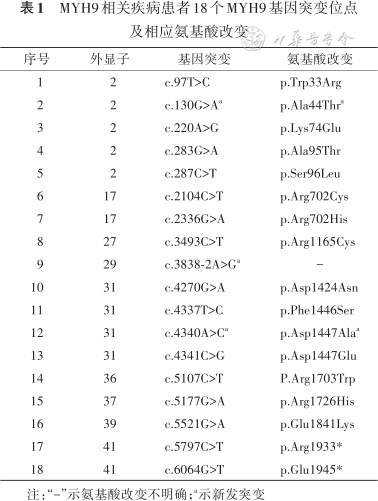
来自35个不相关家系的52例患者进行了基因分析,共发现18个基因突变位点(图1),包括15个错义突变、1个剪切突变、2个终止突变。其中较为常见的突变位点为p.Asp1424Asn(8个家系,12例患者)、p.Arg1933*(5个家系,11例患者)及p.Arg702 His/Cys(5个家系,6例患者),约占所有行基因检测患者的56%(29/52);p.Ser96Leu、Arg1165Cys及p.Glu1841Lys突变属于重现型突变;p.Ala44Thr、p.Asp1447Ala及c.3838-2A>G为新发突变(表1、2)。
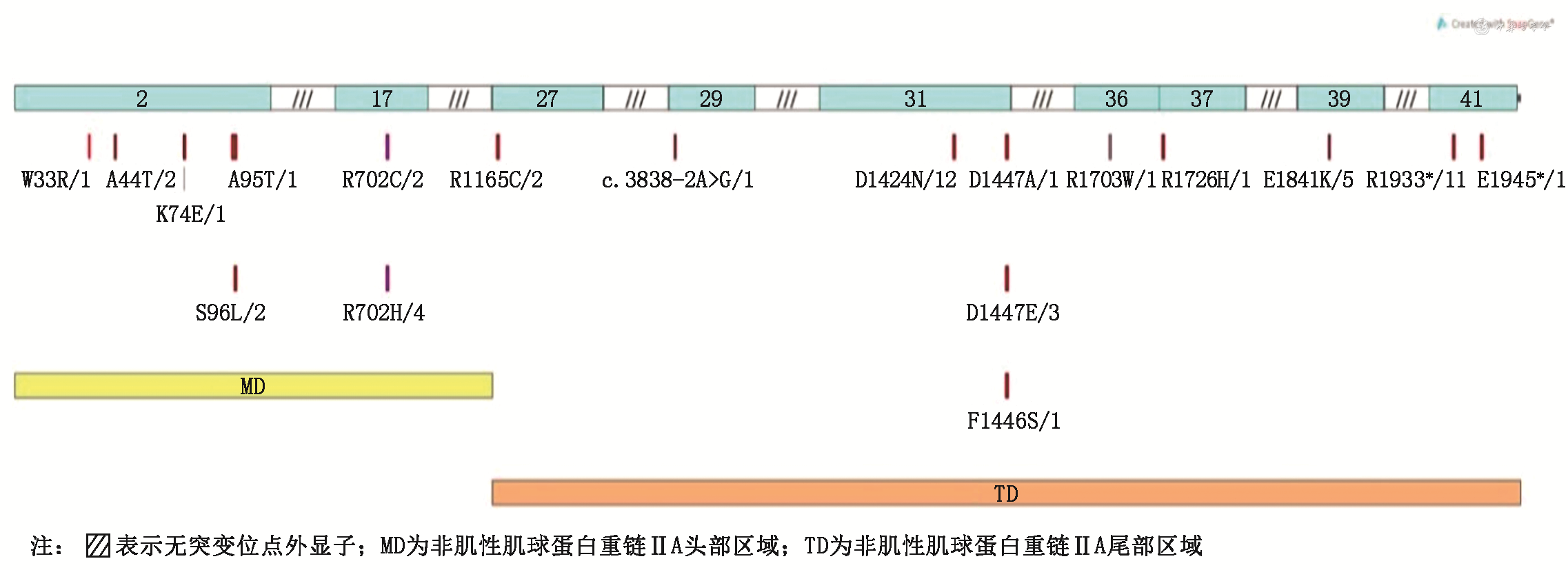
MYH9相关疾病患者18个MYH9基因突变位点及相应氨基酸改变
MYH9相关疾病患者18个MYH9基因突变位点及相应氨基酸改变
| 序号 | 外显子 | 基因突变 | 氨基酸改变 |
|---|---|---|---|
| 1 | 2 | c.97T>C | p.Trp33Arg |
| 2 | 2 | c.130G>Aa | p.Ala44Thra |
| 3 | 2 | c.220A>G | p.Lys74Glu |
| 4 | 2 | c.283G>A | p.Ala95Thr |
| 5 | 2 | c.287C>T | p.Ser96Leu |
| 6 | 17 | c.2104C>T | p.Arg702Cys |
| 7 | 17 | c.2336G>A | p.Arg702His |
| 8 | 27 | c.3493C>T | p.Arg1165Cys |
| 9 | 29 | c.3838-2A>Ga | - |
| 10 | 31 | c.4270G>A | p.Asp1424Asn |
| 11 | 31 | c.4337T>C | p.Phe1446Ser |
| 12 | 31 | c.4340A>Ca | p.Asp1447Alaa |
| 13 | 31 | c.4341C>G | p.Asp1447Glu |
| 14 | 36 | c.5107C>T | P.Arg1703Trp |
| 15 | 37 | c.5177G>A | p.Arg1726His |
| 16 | 39 | c.5521G>A | p.Glu1841Lys |
| 17 | 41 | c.5797C>T | p.Arg1933* |
| 18 | 41 | c.6064G>T | p.Glu1945* |
注:“-”示氨基酸改变不明确;a示新发突变
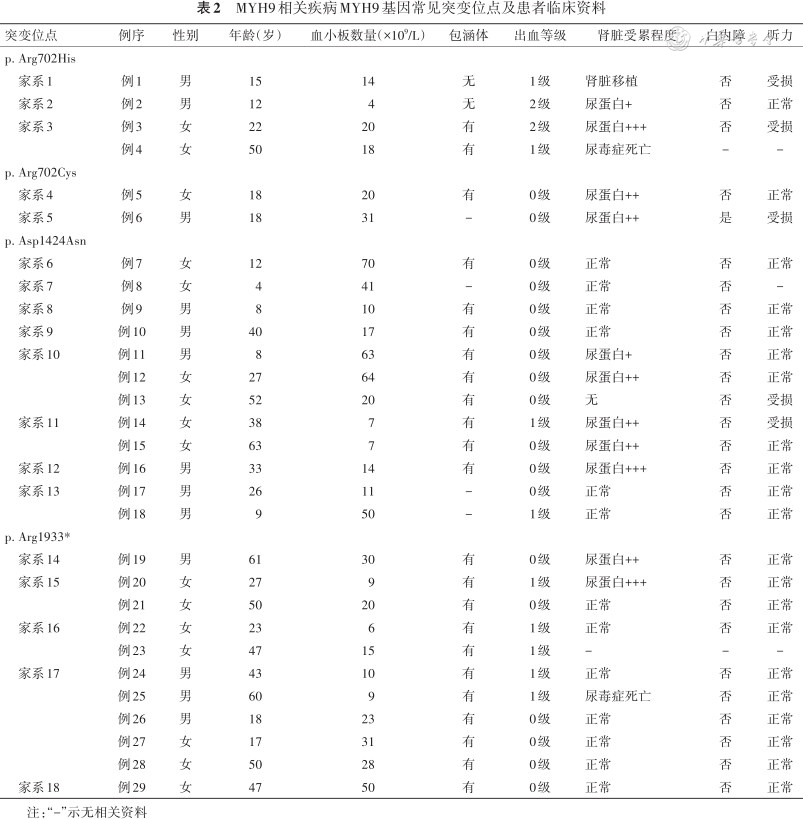
MYH9相关疾病MYH9基因常见突变位点及患者临床资料
MYH9相关疾病MYH9基因常见突变位点及患者临床资料
| 突变位点 | 例序 | 性别 | 年龄(岁) | 血小板数量(×109/L) | 包涵体 | 出血等级 | 肾脏受累程度 | 白内障 | 听力 |
|---|---|---|---|---|---|---|---|---|---|
| p. Arg702His | |||||||||
| 家系1 | 例1 | 男 | 15 | 14 | 无 | 1级 | 肾脏移植 | 否 | 受损 |
| 家系2 | 例2 | 男 | 12 | 4 | 无 | 2级 | 尿蛋白+ | 否 | 正常 |
| 家系3 | 例3 | 女 | 22 | 20 | 有 | 2级 | 尿蛋白+++ | 否 | 受损 |
| 例4 | 女 | 50 | 18 | 有 | 1级 | 尿毒症死亡 | - | - | |
| p. Arg702Cys | |||||||||
| 家系4 | 例5 | 女 | 18 | 20 | 有 | 0级 | 尿蛋白++ | 否 | 正常 |
| 家系5 | 例6 | 男 | 18 | 31 | - | 0级 | 尿蛋白++ | 是 | 受损 |
| p. Asp1424Asn | |||||||||
| 家系6 | 例7 | 女 | 12 | 70 | 有 | 0级 | 正常 | 否 | 正常 |
| 家系7 | 例8 | 女 | 4 | 41 | - | 0级 | 正常 | 否 | - |
| 家系8 | 例9 | 男 | 8 | 10 | 有 | 0级 | 正常 | 否 | 正常 |
| 家系9 | 例10 | 男 | 40 | 17 | 有 | 0级 | 正常 | 否 | 正常 |
| 家系10 | 例11 | 男 | 8 | 63 | 有 | 0级 | 尿蛋白+ | 否 | 正常 |
| 例12 | 女 | 27 | 64 | 有 | 0级 | 尿蛋白++ | 否 | 正常 | |
| 例13 | 女 | 52 | 20 | 有 | 0级 | 无 | 否 | 受损 | |
| 家系11 | 例14 | 女 | 38 | 7 | 有 | 1级 | 尿蛋白++ | 否 | 受损 |
| 例15 | 女 | 63 | 7 | 有 | 0级 | 尿蛋白++ | 否 | 正常 | |
| 家系12 | 例16 | 男 | 33 | 14 | 有 | 0级 | 尿蛋白+++ | 否 | 正常 |
| 家系13 | 例17 | 男 | 26 | 11 | - | 0级 | 正常 | 否 | 正常 |
| 例18 | 男 | 9 | 50 | - | 1级 | 正常 | 否 | 正常 | |
| p. Arg1933* | |||||||||
| 家系14 | 例19 | 男 | 61 | 30 | 有 | 0级 | 尿蛋白++ | 否 | 正常 |
| 家系15 | 例20 | 女 | 27 | 9 | 有 | 1级 | 尿蛋白+++ | 否 | 正常 |
| 例21 | 女 | 50 | 20 | 有 | 0级 | 正常 | 否 | 正常 | |
| 家系16 | 例22 | 女 | 23 | 6 | 有 | 1级 | 正常 | 否 | 正常 |
| 例23 | 女 | 47 | 15 | 有 | 1级 | - | - | - | |
| 家系17 | 例24 | 男 | 43 | 10 | 有 | 1级 | 正常 | 否 | 正常 |
| 例25 | 男 | 60 | 9 | 有 | 1级 | 尿毒症死亡 | 否 | 正常 | |
| 例26 | 男 | 18 | 23 | 有 | 0级 | 正常 | 否 | 正常 | |
| 例27 | 女 | 17 | 31 | 有 | 0级 | 正常 | 否 | 正常 | |
| 例28 | 女 | 50 | 28 | 有 | 0级 | 正常 | 否 | 正常 | |
| 家系18 | 例29 | 女 | 47 | 50 | 有 | 0级 | 正常 | 否 | 正常 |
注:“-”示无相关资料
根据有无出血将患者分为两组,出血组患者血小板数量为(19±3)×109/L,明显少于未出血组的(36±3)×109/L,差异有统计学意义(t=3.64,P<0.001)。根据血小板数量将患者分为两组,血小板数量≤50×109/L的患者中出血等级为0级的有32例[血小板数量(27±2)×109/L],1级有15例[血小板数量(19±3)×109/L],2级有5例[血小板数量(12±3)×109/L],3级有1例(血小板数量7×109/L);血小板数量>50×109/L的患者有13例,无出血表现,差异有统计学意义(统计值Z=-2.687,P=0.007)(表3)。Spearman相关性分析发现血小板数量≤50×109/L的患者组出血与血小板数量呈负相关(r=-0.322,P=0.019)。
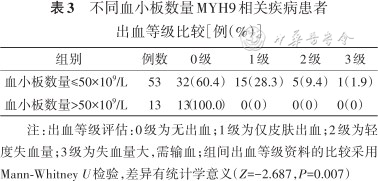
不同血小板数量MYH9相关疾病患者出血等级比较[例(%)]
不同血小板数量MYH9相关疾病患者出血等级比较[例(%)]
| 组别 | 例数 | 0级 | 1级 | 2级 | 3级 |
|---|---|---|---|---|---|
| 血小板数量≤50×109/L | 53 | 32(60.4) | 15(28.3) | 5(9.4) | 1(1.9) |
| 血小板数量>50×109/L | 13 | 13(100.0) | 0(0) | 0(0) | 0(0) |
注:出血等级评估:0级为无出血;1级为仅皮肤出血;2级为轻度失血量;3级为失血量大,需输血;组间出血等级资料的比较采用Mann-Whitney U检验,差异有统计学意义(Z=-2.687,P=0.007)
根据突变位点将患者分为MD突变组和TD突变组。与TD突变组比较,MD突变组血小板数量更少,出血等级更高,差异有统计学意义(Z=-2.295、-2.036,均P<0.05)(表4、5)。同时MD突变组患者在肾脏受累程度、听力受损及白内障等临床表型症状都重于TD突变组,差异均有统计学意义(Z=-2.213、-2.230、-2.812,均P<0.05)(表6、7)。4例患者发生肾功能衰竭,2例携带p.Arg702His突变,2例携带p.Arg1933*及p.Glu1841Lys突变且发病年龄均>50岁(表2)。未见到包涵体的5例患者分别存在c.3838-2A>G(1例)、p.Ser96Leu(1例)、p.Arg1703Trp(1例)及p.Arg702His(2例)突变。
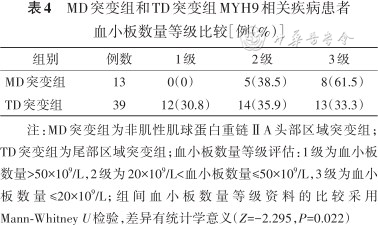
MD突变组和TD突变组MYH9相关疾病患者血小板数量等级比较[例(%)]
MD突变组和TD突变组MYH9相关疾病患者血小板数量等级比较[例(%)]
| 组别 | 例数 | 1级 | 2级 | 3级 |
|---|---|---|---|---|
| MD突变组 | 13 | 0(0) | 5(38.5) | 8(61.5) |
| TD突变组 | 39 | 12(30.8) | 14(35.9) | 13(33.3) |
注:MD突变组为非肌性肌球蛋白重链ⅡA头部区域突变组;TD突变组为尾部区域突变组;血小板数量等级评估:1级为血小板数量>50×109/L,2级为20×109/L<血小板数量≤50×109/L,3级为血小板数量≤20×109/L;组间血小板数量等级资料的比较采用Mann-Whitney U检验,差异有统计学意义(Z=-2.295,P=0.022)
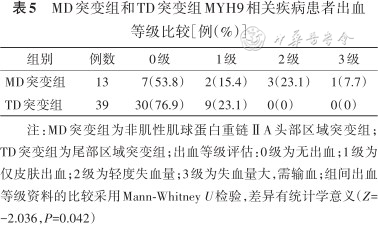
MD突变组和TD突变组MYH9相关疾病患者出血等级比较[例(%)]
MD突变组和TD突变组MYH9相关疾病患者出血等级比较[例(%)]
| 组别 | 例数 | 0级 | 1级 | 2级 | 3级 |
|---|---|---|---|---|---|
| MD突变组 | 13 | 7(53.8) | 2(15.4) | 3(23.1) | 1(7.7) |
| TD突变组 | 39 | 30(76.9) | 9(23.1) | 0(0) | 0(0) |
注:MD突变组为非肌性肌球蛋白重链ⅡA头部区域突变组;TD突变组为尾部区域突变组;出血等级评估:0级为无出血;1级为仅皮肤出血;2级为轻度失血量;3级为失血量大,需输血;组间出血等级资料的比较采用Mann-Whitney U检验,差异有统计学意义(Z=-2.036,P=0.042)
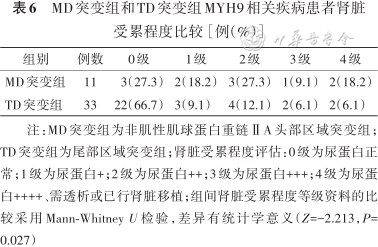
MD突变组和TD突变组MYH9相关疾病患者肾脏受累程度比较[例(%)]
MD突变组和TD突变组MYH9相关疾病患者肾脏受累程度比较[例(%)]
| 组别 | 例数 | 0级 | 1级 | 2级 | 3级 | 4级 |
|---|---|---|---|---|---|---|
| MD突变组 | 11 | 3(27.3) | 2(18.2) | 3(27.3) | 1(9.1) | 2(18.2) |
| TD突变组 | 33 | 22(66.7) | 3(9.1) | 4(12.1) | 2(6.1) | 2(6.1) |
注:MD突变组为非肌性肌球蛋白重链ⅡA头部区域突变组;TD突变组为尾部区域突变组;肾脏受累程度评估:0级为尿蛋白正常;1级为尿蛋白+;2级为尿蛋白++;3级为尿蛋白+++;4级为尿蛋白++++、需透析或已行肾脏移植;组间肾脏受累程度等级资料的比较采用Mann-Whitney U检验,差异有统计学意义(Z=-2.213,P=0.027)
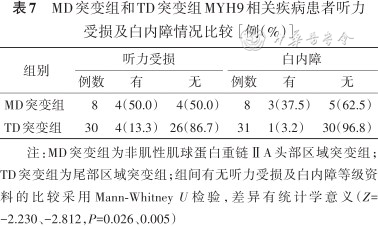
MD突变组和TD突变组MYH9相关疾病患者听力受损及白内障情况比较[例(%)]
MD突变组和TD突变组MYH9相关疾病患者听力受损及白内障情况比较[例(%)]
| 组别 | 听力受损 | 白内障 | ||||
|---|---|---|---|---|---|---|
| 例数 | 有 | 无 | 例数 | 有 | 无 | |
| MD突变组 | 8 | 4(50.0) | 4(50.0) | 8 | 3(37.5) | 5(62.5) |
| TD突变组 | 30 | 4(13.3) | 26(86.7) | 31 | 1(3.2) | 30(96.8) |
注:MD突变组为非肌性肌球蛋白重链ⅡA头部区域突变组;TD突变组为尾部区域突变组;组间有无听力受损及白内障等级资料的比较采用Mann-Whitney U检验,差异有统计学意义(Z=-2.230、-2.812,P=0.026、0.005)
MYH9-RD是一种较常见的遗传性血小板疾病,约98%的MYH9-RD患者存在血小板减少[9],其中80%~90%的患者无自发性出血或只有轻微的皮肤出血,在经历手术、分娩、抗血小板药物治疗等后才有可能发生严重大出血,危及生命的出血更是罕见[3]。本研究结合外周血涂片包涵体及基因测序分析诊断了来自45个家系的66例MYH9患者,血小板减少程度不同,其中约80%的患者血小板数目<50×109/L,血小板聚集功能基本正常,仅30%的患者存在出血症状,且大部分患者表现为轻度的鼻出血、牙龈出血、皮肤瘀点瘀斑,少部分患者因月经量多或阴道出血引起中度贫血。统计分析表明患者的出血程度与血小板数量相关,血小板>50×109/L的患者基本不存在出血的症状。
对于MYH9-RD患者来说,血小板减少是疾病的首发表现,而肾功能不全是该病的严重并发症。约25%的MYH9-RD患者会累及肾脏,表现为进行性蛋白尿,部分患者在几年内迅速进展为终末期肾病[10],而某些病例在生命的后期才出现蛋白尿,且进展较慢[3,11, 12]。本研究中45%患者存在蛋白尿,其中有4例患者发生肾功能衰竭,且2例携带p.Arg702His突变。白内障及听力损伤为晚期疾病的特征,且发病率低于肾脏损伤,其中约20%患者出现听力异常,10%患者存在双侧白内障。因此,需关注MYH9-RD患者肾脏受累情况。
国外研究表明,MYH9-RD患者的临床表现与突变位点具有一定相关性,MD突变时,患者出血较重,伴有严重肾功能不全、神经性耳聋的风险更高,以携带MYH9 Arg702突变的患者临床表现最为严重;TD突变时,患者出血及发生非血液系统症状的风险较低,其中携带MYH9 Arg1933*突变的患者一般不发生肾功能不全或白内障,仅在年老时可能伴发听力障碍[13]。本研究共发现18个基因突变位点,MD突变组患者血小板数量降低更为明显,出血的风险更高,更容易引起非血液症状,如蛋白尿或肾功能不全、听力受损及白内障。国内人群中较为常见的突变位点为p.Asp1424Asn、p.Arg1933*、p.Arg702His/Cys、p.Ser96Leu、p.Arg1165Cys及p.Glu1841Lys,占突变位点的73%。本研究中6例患者出现p.Arg702 His及p.Arg702Cys两类突变,血小板降低明显,均<20×109/L,多数在青少年期即出现明显的蛋白尿,2例患者出现肾功能衰竭,进一步证实Arg702突变致病性较为严重[14]。而p.Asp1424Asn、p.Arg1933*突变发生率最高,患者血小板减少不明显,患者一般在60岁之前伴肾功能不全,但进展程度较轻,多数仅表现为蛋白尿等症状,但本研究中仍有2例患者在晚年发生肾功能衰竭,对于该类突变需要引起重视。
本研究中41%的MYH9-RD患者为散发病例,缺少家族史,高于国际35%的报道[8],这也是容易造成漏诊的原因之一。重视MYH9-RD患者的实验室特征有利于提高该病的诊断率。本研究中所有患者均存在大血小板减少,既往研究发现MYH9-RD患者的平均血小板直径为4.5 μm,而健康对照组的平均血小板直径为2.6 μm[3]。而中性粒细胞蓝色包涵体是MYH9-RD 患者的特征性临床表现,可见于本研究中91%的患者,需要注意的是不同类型突变可产生不同形式的包涵体[14],从而导致检测人员在认知上存在偏差。而免疫荧光分析粒细胞 NMMHC-ⅡA 包涵体能显示绝大多数包涵体,但操作复杂,需要专门人员和仪器。随着NGS的普及,可以直接对MYH9基因进行测序,大大提高了该病的诊断效率。因此,外周血细胞形态筛查联合基因测序在诊断MYH9-RD方面具有非常好的敏感性和特异性,及早进行有利于减少误诊及不恰当的治疗。
血小板减少是MYH9-RD早期的表现,但患者一般出血较轻,不需要特殊处理,对于严重出血患者及围手术期预防出血的主要治疗方式为输注血小板以及人工刺激血小板生成[15]。促血小板生成素受体激动剂(thrombopoietin receptor agonist,TPO-RA)如艾曲泊帕和罗米司亭可有效提升血小板数量[16]。目前关于肾病的治疗报道较少,一些研究表明阻断肾素-血管紧张素系统可以有效降低蛋白尿水平[17];但进展为终末期肾病时,只能通过透析和肾脏移植来治疗[18, 19],因此在减缓肾功能不全方面迫切需要新的治疗方案。听力障碍的患者,使用助听器或人工耳蜗移植有助于听力恢复[20, 21]。
综上所述,MYH9-RD表现为常染色体显性遗传,发病率并不低。临床上应提高对该类疾病的认识,做到早期诊断、早期预防严重并发症,这可以减少患者经济及心理负担。同时应加强产前诊断,减少该疾病的发生。本研究通过分析MYH9-RD临床特征及基因突变情况,为MYH9-RD这一常见的遗传性血小板减少症的诊断及治疗提供借鉴。
高歌阳, 曹丽娟, 余自强, 等. MYH9相关疾病临床表型及基因突变特征分析[J]. 中华医学杂志, 2023, 103(37): 2964-2970. DOI: 10.3760/cma.j.cn112137-20230328-00496.
所有作者声明不存在利益冲突

























