
本文报告1例早产儿先天性长QT综合征,患儿于胎儿期即发现胎心不齐,生后出现严重心律失常,心率55~110次/min,24 h动态心电图示QT间期延长、尖端扭转型心律失常、二度Ⅱ型房室传导阻滞,基因检测发现KCNH2基因变异,生后10 d家长放弃治疗后死亡。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿女,生后2 h因“低血糖”收入院。患儿系第3胎第2产,胎龄36+1周剖宫产娩出,出生体重2 920 g,出生史无特殊,生后监测2次血糖,分别为1.9 mmol/L、1.2 mmol/L,无抽搐发作。母亲既往人工流产1次;此次孕期规律产检,孕35+6周产检时发现胎儿心律不齐,胎心基线110~145次/min,收入院,予地塞米松4次促进胎肺成熟,入院后2 d行剖宫产。胞姐11岁,体健。入院查体:心率110次/min,心律不齐,心音正常。腹部平坦,肌张力正常,原始反射存在。入院后复查末梢血糖2.1 mmol/L,血气分析、血电解质无异常,心脏超声及心肌肌钙蛋白均正常。24 h动态心电图:QT间期延长、尖端扭转型心律失常、二度Ⅱ型房室传导阻滞。入院后予葡萄糖静脉输注维持血糖,入院第2天予25%硫酸镁0.2 ml/kg静脉输注1次,予果糖二磷酸钠0.16 g/(kg·d)营养心肌,心律失常无改善;第3天加用异丙肾上腺素0.2 μg/(kg·min),第5、6天提高至0.7 μg/(kg·min),第7、8天下调至0.5 μg/(kg·min);第4天出现发热、腹胀,诊断新生儿坏死性小肠结肠炎(necrotizing enterocolitis,NEC),予头孢噻肟钠、哌拉西林他唑巴坦加强抗感染等治疗。由于家属拒绝进一步治疗,生后10 d患儿心脏搏动、自主呼吸微弱,家属放弃治疗后死亡。生后第6天取血进行基因检测,采用目标区域捕获+高通量测序法,24 d后结果回报:患儿KCNH2基因存在一处杂合变异c.1478A>G,导致氨基酸改变Tyr493Cys,该变异为新发变异,父母不携带该变异。
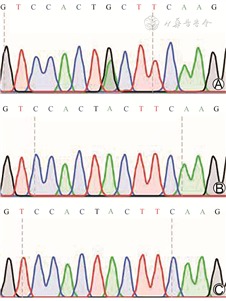
先天性长QT综合征(long QT syndrome,LQTs)是一种恶性心律失常,为常染色体隐性遗传病[1],发病率尚无准确的流行病学数据,患病率约为1/2 500,平均发病年龄为14岁[2]。目前研究表明,LQTs与17种不同基因变异有关,其中KCNQ1、KCNH2和SCN5A基因最具代表性[3]。先天性LQTs的发病机制是心脏离子通道改变导致的恶性心律失常,尤其是尖端扭转型心动过速,容易引起晕厥、癫痫样抽搐和心跳骤停、猝死,在新生儿期发病时病死率极高[3]。此外由于心律不齐、血压不稳定导致各脏器血流灌注不足,肠道缺血、功能受损尤为严重,特别是早产儿,可在生后1周内发生严重NEC而死亡[4]。2019年Crotti 等[4]报道了3例在新生儿期诊断LQTs的儿童,分别在生后8 d、6个月和5岁死于埋藏式心律转复除颤器植入并发症、心脏手术后心力衰竭和感染合并低血糖。本例患儿宫内即发现心律不齐,生后心电图提示存在严重的恶性心律失常(尖端扭转、房室传导阻滞等多种类型),基因检测发现KCNH2基因变异,最终因合并NEC,家长放弃治疗死亡。
先天性LQTs治疗效果极差,在早产儿中极为罕见、致死率极高[5]。在大多数患者中,β 受体阻滞剂仍然是预防危及生命的心律失常的主要治疗方法;其他治疗方法包括起搏器、植入式心脏复律除颤器、左侧颈交感神经切除术、钠通道阻断药物和改变生活方式。本例患儿进行了硫酸镁、异丙肾上腺素对症支持治疗,但效果不佳,如能早期植入心脏起搏器,或许可增加存活概率。
陈蓉蓉, 王晓康, 张小华. 早产儿长QT综合征1例[J]. 中华新生儿科杂志, 2023, 38(11): 691-692. DOI: 10.3760/cma.j.issn.2096-2932.2023.11.013.
所有作者声明无利益冲突

























