
遗传性表面活性物质功能障碍疾病是表面活性物质合成和代谢过程中的相关基因变异引起的一组疾病,是儿童间质性肺疾病的重要病因。随着基因测序技术的广泛应用,本病诊断门槛降低,但识别临床线索以及治疗管理的能力仍有待加强。为提高儿科医师对本病的认识,促进早期诊断和规范治疗,改善患儿预后,特组织专家制定本共识。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
遗传性表面活性物质功能障碍疾病(genetic disorders of surfactant dysfunction,GDSD)是由基因变异引起肺表面活性物质(pulmonary surfactant,PS)功能异常所致的疾病,表现为新生儿呼吸窘迫综合征(respiratory distress syndrome,RDS)和/或儿童间质性肺疾病(interstitial lung disease,ILD)。PS由Ⅱ型肺泡上皮细胞(alveolar type Ⅱ epithelial cells,ATⅡ)合成并分泌到肺泡表面,具有降低肺泡表面张力、防止呼气末肺萎陷的生理作用。在PS合成和代谢途径中,一些重要蛋白的编码基因发生致病性变异,可导致PS的合成和代谢过程受阻,引起GDSD。目前已知与GDSD有关的基因主要有SFTPB、SFTPC、ABCA3、NKX2.1、CSF2RA和CSF2RB,分别编码表面活性蛋白(surfactant protein,SP)-B、SP-C、ATP结合盒A家族成分3 (ATP binding cassette subfamily A member 3,ABCA3)、甲状腺转录因子(thyroid transcription factor, TTF)-1以及粒细胞-巨噬细胞集落刺激因子(granulocyte-macrophage colony-stimulating factor,GM-CSF)受体α和β亚基。
PS由重量占90%的磷脂和占10%的蛋白质组成,其中二棕榈酰磷脂酰胆碱以及SP-B、SP-C是PS发挥表面活性作用的重要成分。SP-B与SP-C是小分子疏水性蛋白,通过调节磷脂分子层在液气表面的结构和分布,使PS在肺泡表面扩散形成单层覆膜,从而调节肺泡表面张力。此外还有2种表面活性蛋白,SP-A和SP-D是亲水性蛋白,主要发挥固有免疫和免疫调节的生理功能。
PS在AT Ⅱ细胞内合成。在AT Ⅱ细胞胞质中的溶酶体来源细胞器-板层小体内,SP-B、SP-C和磷脂组装合成PS[1]。磷脂的转运依赖于板层小体界膜上的跨膜蛋白ABCA3。板层小体的结构稳定也依赖功能正常的ABCA3和SP-B。SP-B、SP-C和ABCA3的蛋白合成受NKX2.1基因编码的TTF-1调控。通过胞吐作用,AT Ⅱ细胞将合成的PS分泌到肺泡发挥生理作用。
PS的代谢有2种途径,一部分被ATⅡ细胞再摄取和利用,另一部分被肺泡巨噬细胞吞噬和降解。肺泡巨噬细胞的分化成熟需要GM-CSF结合巨噬细胞表面的CSF受体,进一步激活下游信号通路。GM-CSF含量减少或CSF受体缺陷,将导致肺泡巨噬细胞功能异常,PS清除障碍,最终引起脂蛋白在肺泡沉积[2]。
PS合成和代谢过程见图1。
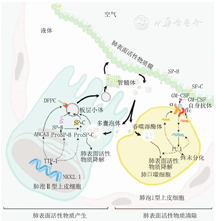
注:DPPC:二棕榈酰磷脂酰胆碱;SP-B:表面活性蛋白B;SP-C:表面活性蛋白C;ProSP-B:SP-B前体蛋白;ProSP-C:SP-C前体蛋白;ABCA3:ATP结合盒A家族成分3;NKX2.1:NK2同源框1;TTF-1:甲状腺转录因子1;GM-CSF:粒细胞-巨噬细胞集落刺激因子 DPPC:dipalmitoyl phosphatidylcholine;SP-B:surfactant protein B;SP-C:surfactant protein C;ProSP-B:prosurfactant protein B;ProSP-C:prosurfactant protein C;ABCA3:ATP binding cassette subfamily A member 3;NKX2.1:NK2 homeobox 1;TTF-1:thyroid transcription factor 1;GM-CSF:granulocyte-macrophage colony-stimulating factor
PS的合成和代谢途径中,上述重要蛋白的编码基因发生变异,将引起PS功能障碍,导致GDSD。目前已经明确的致病基因主要有SFTPB、SFTPC、ABCA3、NKX2.1以及CSF2RA和CSF2RB,基因型和表型特征以及致病机制见表1。SP-A的编码基因SFTPA2变异可引起成人肺纤维化[3],在儿童罕见。
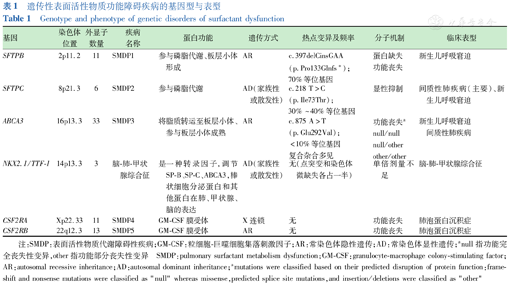
遗传性表面活性物质功能障碍疾病的基因型与表型
Genotype and phenotype of genetic disorders of surfactant dysfunction
遗传性表面活性物质功能障碍疾病的基因型与表型
Genotype and phenotype of genetic disorders of surfactant dysfunction
| 基因 | 染色体位置 | 外显子数量 | 疾病名称 | 蛋白功能 | 遗传方式 | 热点变异及频率 | 分子机制 | 临床表型 |
|---|---|---|---|---|---|---|---|---|
| SFTPB | 2p11.2 | 11 | SMDP1 | 参与磷脂代谢、板层小体形成 | AR | c.397delCinsGAA(p.Pro133Glnfs*);70%等位基因 | 蛋白缺失功能丧失 | 新生儿呼吸窘迫 |
| SFTPC | 8p21.3 | 6 | SMDP2 | 参与磷脂代谢 | AD(家族性或散发性) | c.218 T>C (p.Ile73Thr);30%~40%等位基因 | 显性抑制 | 间质性肺疾病(主要)、新生儿呼吸窘迫 |
| ABCA3 | 16p13.3 | 33 | SMDP3 | 将脂质转运至板层小体、参与板层小体成熟 | AR | c.875 A>T(p.Glu292Val);<10%等位基因复合杂合多见 | 功能丧失anull/nullnull/otherother/other | 新生儿呼吸窘迫间质性肺疾病 |
| NKX2.1/TTF-1 | 14p13.3 | 3 | 脑-肺-甲状腺综合征 | 是一种转录因子,调节SP-B、SP-C、ABCA3,棒状细胞分泌蛋白和其他蛋白在肺、甲状腺、脑的表达 | AD(家族性或散发性) | 无(点突变和染色体微缺失各占一半) | 单倍剂量不足 | 脑-肺-甲状腺综合征 |
| CSF2RA | Xp22.33 | 11 | SMDP4 | GM-CSF膜受体 | X连锁 | 无 | 功能丧失 | 肺泡蛋白沉积症 |
| CSF2RB | 22q12.3 | 13 | SMDP5 | GM-CSF膜受体 | AR | 无 | 功能丧失 | 肺泡蛋白沉积症 |
注:SMDP:表面活性物质代谢障碍性疾病;GM-CSF:粒细胞-巨噬细胞集落刺激因子;AR:常染色体隐性遗传;AD:常染色体显性遗传;anull指功能完全丧失性变异,other指功能部分丧失性变异 SMDP:pulmonary surfactant metabolism dysfunction;GM-CSF:granulocyte-macrophage colony-stimulating factor;AR:autosomal recessive inheritance;AD:autosomal dominant inheritance;amutations were classified based on their predicted disruption of protein function:frameshift and nonsense mutations were classified as"null"whereas missense,predicted splice site mutations,and insertion/deletions were classified as"other"
GDSD的共同表现为足月新生儿或晚期早产儿发生RDS和/或儿童期,甚至成年后起病的ILD。在不同年龄段,以及不同致病基因引起的GDSD,临床表现有所不同。
GDSD在新生儿期表现为RDS,主要是足月儿或晚期早产儿发生RDS,偶尔也见于早期早产儿。与早产儿肺发育不成熟引起的RDS不同,GDSD引起的RDS,其严重程度、治疗反应和预后与致病基因类型密切相关。绝大部分的SFTPB变异以及大约60%的ABCA3变异,引起严重新生儿RDS,表面活性物质替代治疗无效,常导致死亡或需要肺移植[4]。大约40%的ABCA3变异,以及大部分SFTPC、NKX2.1变异引起的RDS,在短期的呼吸支持和表面活性物质替代治疗后可能完全缓解一段时间,直到ILD发病;一些则只有部分缓解,持续咳嗽、气促、低氧血症等慢性呼吸道症状,直到胸部影像证实ILD。NKX2.1变异引起脑-肺-甲状腺综合征,除了RDS,在新生儿期还可以出现喂养困难、肌张力低下、甲状腺功能低下等临床表现或实验室检查异常[5]。
根据国外数据,表现为新生儿RDS的GDSD以检出ABCA3变异最常见[5]。
GDSD在婴幼儿期主要表现为ILD,即慢性咳嗽、气促、低氧血症,胸部影像出现双肺弥漫性间质性/实质性病变,常伴有生长发育落后。出生后第1年起病的ILD往往与GDSD有关[1,2],患儿常有新生儿期RDS病史。
婴幼儿期的GDSD以检出SFTPC变异最常见[6]。SFTPC基因外显率和表现度可变,表型个体差异大,即使致病位点完全相同,临床的轻重和预后也可以不同[7,8]。根据国外报道,3/4的病例在1岁前起病,超过一半出现慢性呼吸功能不全和生长发育落后,呼吸系统症状还表现为慢性咳嗽和反复呼吸道感染,可出现杵状指/趾,10%的病例可无明显症状[9]。我国SFTPC变异患儿的临床表现同样如此,有文献报道7例患儿中的5例起病年龄<1岁,均表现为咳嗽、气促、呼吸困难、低氧血症,伴生长发育落后,其中4例有杵状指/趾[10]。
ABCA3变异的临床表现和严重程度与变异位点的类型及其对蛋白表达的影响程度有关。如果2个等位基因都是功能丧失性变异(如无义变异和移码变异),将导致类似SFTPB变异引起致死性RDS;至少1个等位基因是相对温和的变异时(如错义变异),导致ABCA3功能降低而非完全丧失,多表现为暂时性新生儿RDS和/或儿童ILD,但往往比SFTPC变异的症状更重、治疗反应更差[4,11]。国内报道的ABCA3变异病例临床表现和严重程度个体间差异大,可表现为轻度呼吸道症状,或经治疗后症状和肺部CT有所改善呈慢性肺病状态[12,13],或治疗无反应最终行肺移植[14,15]。具有相同的ABCA3变异位点的不同患者,临床表型严重程度可以不同[12]。
NKX2.1变异的典型表现为"脑-肺-甲状腺综合征",即肺部症状、甲状腺功能低下以及舞蹈病、共济失调、发育落后和肌张力减退等神经系统症状;一部分NKX2.1变异病例仅累及呼吸系统,临床表现除了ILD,还可表现为反复呼吸道感染、重症感染[16,17]。在国内报道的4例NKX2.1变异中,诊断年龄从新生儿至15岁,2例足月儿、2例32周早产儿;其中3例新生儿期有RDS,2例ILD存在氧依赖,2例反复肺部感染;4例均有甲状腺功能低下;神经系统表现为头围小,肌张力低下,精神、运动、语言和生长发育落后;2例牙齿发育不良,伴IgG水平下降,这2例为染色体微缺失。对于染色体微缺失引起的"脑-肺-甲状腺综合征",可能合并其他基因缺陷的表型,如牙齿发育不良、体液免疫缺陷等[18,19,20]。
除了小部分SFTPC变异在儿童和青少年期起病以外,这个年龄段的GDSD多为婴幼儿期ILD的延续,表现为慢性肺病持续状态。婴幼儿期的ILD经治疗后,症状不同程度缓解,可无临床症状,或需要长期的抗炎治疗以维持较轻的慢性呼吸道症状,部分患儿可能需要长达数年的吸氧等呼吸支持,甚至最终需要肺移植。即使已无症状,患儿的胸部影像常持续存在双肺弥漫性间质性病变。SFTPC变异患儿胸部影像有逐渐进展为肺纤维化的趋势,临床症状可能在某个阶段再现。ABCA3变异年长儿还可出现漏斗胸等胸廓畸形[13]。
CSF2RA和CSF2RB变异引起遗传性肺泡蛋白沉积症(pulmonary alveolar proteinosis, PAP)[21,22],轻者可无明显症状,重者进行性呼吸衰竭。国内报道1例3岁3月龄患儿,CSF2RA变异引起PAP,表现为咳嗽、活动耐力下降、呼吸困难,经抗感染、甲泼尼龙抗炎以及丙种球蛋白免疫调节等治疗仍无法拔管撤离呼吸机,经多次全肺灌洗等治疗后有所好转[23]。
主要基于RDS或ILD的临床表现和胸部CT影像,通过基因检测明确诊断GDSD。肺组织病理学用于基因检测不能明确诊断,或病情进展快急需获得诊断依据的患者。支气管肺泡灌洗液(bronchoalveolar lavage fluid,BALF)检查常用于排除感染等其他导致弥漫性肺病的病因以及辅助诊断PAP;肺功能主要用于疾病的随访监测;心脏彩超用于评估有无肺动脉高压。以下主要介绍胸部CT、基因检测和肺组织病理特征。
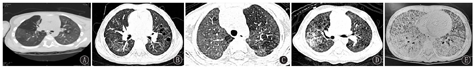
高分辨率CT(high-resolution computed tomography,HRCT)对发现肺间质性病变的敏感性高,GDSD的胸部HRCT常见表现为肺实质弥漫性磨玻璃影以及小叶内间质和小叶间隔增厚、网格影、囊泡影等间质性病变。在病程早期,以肺磨玻璃影常见;抗炎治疗后症状改善,磨玻璃影逐渐吸收,但随着病程推移小叶间隔增厚、网格影和囊泡影等间质性病变更加明显,最终可能出现肺纤维化[7,27,28,29]。ABCA3变异患者随访过程中可能出现漏斗胸[13,30]。CSF2RA和CSF2RB变异引起的PAP,典型肺部CT影像为"铺路石征",即在磨玻璃影的背景上,叠加小叶内间质和小叶间隔增厚等间质病变。
不同基因型GDSD的胸部CT见图2(GDSD的胸部CT影像有共同特征,通过CT表现难以区分不同基因型)。
GDSD是单基因病,基因检测是首选诊断方法[31]。新一代测序(next-generation sequencing,NGS)已广泛应用,目前临床常选择全外显子测序;根据对拟诊疾病的把握度也可选择特定疾病的基因靶向测序(gene panel);全基因组测序可获得包括内含子在内更全面的遗传信息,但解读报告的难度也随之增加。由于NGS检测费用较高,目前临床常选择对先证者进行NGS检测,如检出可疑致病位点,再对家系3人(患儿和父母)的样本进行一代测序验证。如有条件对家系3人都进行NGS检测,更有利于数据分析和致病位点的判定。
GDSD的致病基因、常见变异位点等遗传信息见表1。基因报告一般参照美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)指南[32]对检测结果进行解读,变异位点的临床意义评级有致病、可能致病、临床意义不明确、可能良性、良性5种情况。检出致病的变异位点,具有诊断意义。可能致病代表这个变异有90%~99%的可能性导致蛋白功能异常。检出可能致病的变异位点,结合符合GDSD的临床表现可建立诊断。Clinvar数据库收录的、根据ACMG指南评级为致病和可能致病的GDSD单核苷酸变异位点见附录A。如报告未检出有关的变异位点,还需要注意拷贝数变异、内含子变异等常规全外显子测序可能遗漏的情况,进一步与基因测序公司技术人员深入沟通或转诊至遗传咨询门诊。
基因测序只能检出60%~75%的GDSD[33],基因检测结果阴性或病情快速进展来不及等待基因测序结果的患者,仍需要肺活检和组织病理学检查。首选经胸腔镜肺活检;开胸手术肺活检创伤较大;经支气管透壁肺活检可能无法获得足够的肺组织样本,阳性率低[34];经支气管冷冻肺活检可获得较大的组织样本[35],但应用于儿童技术难度较大。参照指南[36]加工和处理肺组织标本,并留取戊二醛固定的标本以便必要时进行电子显微镜检查。专门的免疫组织化学检查可能提供更多有用信息,目前主要在研究实验室中使用。
光学显微镜下,GDSD的组织病理学特征为肺间质炎症和纤维化,常见AT Ⅱ细胞增生、肺间质增宽、肺泡腔内存在泡沫状肺泡巨噬细胞和数量不等的颗粒状蛋白物质(图3)。脱屑型间质性肺炎(desquamative interstitial pneumonitis,DIP)、婴儿期慢性肺炎(chronic pneumonitis of infancy,CPI)、非特异性间质性肺炎(non-specific interstitial pneumonia,NSIP)、PAP等肺组织病理类型提示GDSD。单一病例有可能观察到几种病理模式。国外一组多中心、小于2岁的症状性肺病患儿的肺活检病理资料显示,SFTPC变异最常见病理类型为CPI[37],特点为肺泡间隔增厚、间充质细胞增生而非炎性浸润,肺泡腔内可有数量不同的含蛋白成分物质[38];ABCA3变异的病理类型主要为PAP、DIP和NSIP,其中PAP主要见于小婴儿[37,39],DIP更常见于表现为ILD的较年长儿童[30,40]。寻常型间质性肺炎(usual interstitial pneumonia,UIP)是成人ILD常见的病理类型,在CT上表现为肺纤维化。UIP在儿童罕见,但有病例报道ABCA3[25]以及SFTPC[26]变异患者的肺组织病理表现为UIP。NKX2.1变异的肺组织病理可见小叶间隔炎症、纤维化,可伴不同程度的肺组织结构异常,如肺泡腔增大、肺泡结构简单化[16]。CSF2RA和CSF2RB变异引起PAP[21,22]。根据组织病理学表现无法鉴别GDSD的遗传学病因[37]。
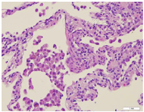
注:肺泡腔部分扩张,腔内可见少许脱落的上皮细胞、红细胞及巨噬细胞,肺间隔增宽,可见淋巴细胞、巨噬细胞浸润,小叶间隔增厚。SP-B(+),SP-C(-) The alveolar spaces are partially dilated,with a few sloughed epithelial cells,red blood cells,and macrophages presenting in the cavity.The lung interstitium is widened,with lymphocytes and macrophages infiltration,as well as interlobular septal thickening.SP-B(+),SP-C(-)
电子显微镜下,ABCA3、SFTPB和NKX2.1变异患者可见AT Ⅱ细胞的板层小体结构异常。ABCA3变异的板层小体较小且密度较高,可见偏心位包涵体、2个或多个板层小体融合,可有部分形态正常[30];SFTPB变异的板层小体结构紊乱且层次不良,存在泡状包涵体[41];NKX2.1变异可见板层小体内不同大小、均质和非均质的致密体及其融合,可有部分形态正常[16]。SFTPC变异患者的板层小体形态可正常,或形成大的板层小体。板层小体出现以上特征性异常支持GDSD诊断。
BALF检查对PAP具有诊断意义:特征性表现为浑浊的乳白色液体,且其浑浊度随连续灌洗而逐渐下降(图4);光学显微镜检查沉积物可见大量无定形、过碘酸-雪夫(PAS)染色呈明亮阳性的脂蛋白性物质,也可能见到较大的泡沫样巨噬细胞;BALF细胞分类计数以巨噬细胞为主,无炎症细胞。CSF2RA和CSF2RB变异引起遗传性PAP,但还有其他基因变异以及原发和继发性病因可引起PAP,因此BALF确诊PAP之后仍需要进行基因检测。
注:支气管肺泡灌洗液呈牛奶样,静置后沉淀,多次灌洗后逐渐转清亮(从右至左) The bronchoalveolar lavage fluid appears milky,with a thick settling layer of sediment if lay standing.It gradually turns clear after repeat lavage(from right to left)
除了诊断PAP以外,支气管镜和BALF检查主要用于感染、嗜酸细胞性肺炎、外源性过敏性肺泡炎等其他引起双肺弥漫性病变的病因鉴别诊断。实验室研究表明,BALF中的SP-B水平降低可能有助于筛查SFTPB变异,SP-C水平降低可见于SFTPC、SFTPB、NKX2.1或ABCA3变异[42],目前尚未用于临床检测。
诊断GDSD的临床线索:(1)足月儿或晚期早产儿发生RDS;(2)早产儿RDS,对表面活性物质替代治疗效果差,或持续氧依赖不符合早产儿支气管肺发育不良的自然病程;(3)新生儿期发生RDS,好转后持续存在呼吸道症状;(4)ILD的临床表现,如慢性咳嗽、气促、呼吸困难、运动不耐受、低氧血症等;(5)体格检查出现吸气性三凹征、细湿啰音、异常呼吸音(附加音或爆裂音)、生长发育落后、杵状指/趾、胸廓畸形等;(6)指脉氧监测持续异常或反复血气分析发现不明原因低氧血症。
存在以上临床线索,和/或胸部CT表现为弥漫性磨玻璃影、小叶间隔增厚、网格影、囊泡影等,初步检查排除其他引起弥漫性肺病的病因后,进行基因检测。GDSD诊断流程见图5。
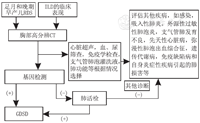
注:RDS:呼吸窘迫综合征;ILD:间质性肺疾病;GDSD:遗传性表面活性物质功能障碍疾病 RDS:respiratory distress syndrome;ILD:interstitial lung disease;GDSD:genetic disorders of surfactant dysfunction
(1)根据临床线索+检出ACMG评级为致病、可能致病的基因变异可做出基因诊断;(2)对于基因检测结果阴性者,必要时进一步行肺活检。临床线索+符合GDSD的肺组织病理类型,和/或电镜下AT Ⅱ细胞特征性板层小体结构异常,可做出病理诊断。GDSD的临床-放射-病理特点见表2。

遗传性表面活性物质功能障碍疾病的临床-放射-病理特点
Clinical,radiologic and pathologic features of genetic disorders of surfactant dysfunction
遗传性表面活性物质功能障碍疾病的临床-放射-病理特点
Clinical,radiologic and pathologic features of genetic disorders of surfactant dysfunction
| 临床特征 | SFTPB变异 | SFTPC变异 | ABCA3变异 | NKX2.1/TTF-1变异 | CSF2RA/CSF2RB变异 |
|---|---|---|---|---|---|
| 起病年龄 | 出生后 | 各个年龄段,婴儿期多见 | 出生后和婴儿期多见 | 各个年龄段,出生后和婴儿期多见 | 婴幼儿期/儿童期 |
| 疾病转归 | 致死性(出生后3~6个月死亡) | 轻重不一/慢性病程 | 致死性/慢性病程 | 轻重不一/慢性病程 | 慢性病程 |
| 临床表现 | |||||
| 新生儿期 | |||||
| 足月儿呼吸窘迫综合征 | 绝大部分病例 | 部分病例 | 部分病例 | 部分病例 | 无 |
| 婴幼儿期 | |||||
| 气促 | + | + | + | + | |
| 活动耐受力下降 | + | + | + | + | |
| 反复呼吸道感染 | + | + | + | + | |
| 慢性咳嗽 | + | + | + | + | |
| 生长发育落后 | + | + | + | + | |
| 继发胸廓畸形 | + | + | - | + | |
| 其他 | - | - | 甲状腺功能减低 | - | |
| 肌力减低、舞蹈症 | |||||
| 高分辨率CT | |||||
| 双肺弥漫磨玻璃影 | + | + | + | + | + |
| 支气管充气征 | + | + | + | - | - |
| 肺囊泡 | - | + | + | + | - |
| 肺气肿 | - | + | + | + | - |
| 支气管扩张 | - | + | - | + | - |
| 铺路石征 | - | - | + | - | + |
| 小叶间隔增厚 | - | + | + | + | + |
| 肺泡结构扭曲 | - | + | + | + | - |
| 病理类型 | PAP、DIP | CPI、DIP、NSIP | NSIP、DIP、PAP、CPI | DIP、NSIP、CPI | PAP |
| 病理(电镜) | 低倍:大LBs、磷脂层消失、内质网肿胀 | 低倍:细胞质中可见密度增高物质,肺泡腔内见大量异质性物质(未成熟的蛋白质)及巨噬细胞 | 低倍:LBs正常/变小 | 低倍:小LBs且密度变高 | 低倍:肺泡腔内大量异常表面活性蛋白 |
| 高倍:失去同心圆结构的LBs,大量MVBs | 高倍:内质网肿胀,可形成同心圆状LBs,大LBs且形状不规则,磷脂分布异常 | 高倍:小LBs且密度变高、可融合、形成煎蛋样的LBs、磷脂层消失 | 高倍:LBs结构异常,失去同心圆结构 | 高倍:肺泡腔中泡沫细胞内可见大量异常LBs和脂滴 | |
| 支气管肺泡灌洗液 | SP-B缺乏,ProSP-C增加 | SP-C缺乏,ProSP-C降低 | SP-B、SP-C降低 | SP-B、SP-C、ABCA3降低 | 可见泡沫细胞,PAS反应及油红"O"染色(+) |
| 治疗 | 呼吸支持,肺移植 | 糖皮质激素、羟氯喹等,吸氧等支持治疗,必要时肺移植 | 糖皮质激素、羟氯喹等,吸氧等支持治疗,必要时肺移植 | 糖皮质激素、羟氯喹等,补充甲状腺素,吸氧等支持治疗,必要时肺移植 | 全肺灌洗,造血干细胞移植,肺移植有争议 |
注:PAP:肺泡蛋白沉积症;DIP:脱屑性间质性肺炎;CPI:婴儿慢性肺炎;NSIP:非特异性间质性肺炎;LBs:板层小体;MVBs:多囊泡体;PAS:过碘酸-雪夫 PAP:pulmonary alveolar proteinosis;DIP:desquamative interstitial pneumonitis;CPI:chronic pneumonitis of infancy;NSIP:non-specific interstitial pneumonia;LBs:lamellar bodies;MVBs:multivesicular bodies;PAS:periodic acid-Schiff
GDSD是婴儿期特有的ILD中的主要疾病。对于婴儿期起病的ILD,在送检基因前应根据我国儿童ILD病因诊断程序[43],先除外环境暴露相关的ILD、系统性疾病相关的ILD以及肺泡结构紊乱相关的ILD。
(1)新生儿期起病的GDSD主要表现为新生儿RDS,需要与早产相关的新生儿RDS、宫内感染引起的急性RDS、严重先天性心脏病、先天性肺发育异常、原发性纤毛运动障碍等疾病鉴别。
(2)婴儿期起病的GDSD主要表现为ILD,需要与感染、吸入性肺炎、外源性过敏性肺泡炎、支气管肺发育不良、弥漫性肺泡出血综合征、遗传代谢病、自身免疫和自身炎症性疾病引起的肺损害等疾病鉴别。
纠正低氧血症,通过吸氧、呼吸支持使脉搏血氧饱和度(SpO2)不低于0.92,如果合并肺动脉高压应达到0.94以上。鼻导管吸氧是最常用的儿童家庭氧疗方式,最大氧流量不超过2 L/min,输出的氧浓度范围为0.22~0.95。指导家长正确的鼻导管固定方法,避免损伤皮肤,通常每周更换一次鼻导管。年长儿童可使用面罩吸氧。加温加湿高流量吸氧、呼吸机可提供更高浓度的氧气。家庭氧疗时,避免在家中吸烟,避免在氧源附近使用明火、油或其他燃料。
超过一半的GDSD患儿生长落后,注意监测身高、体重生长曲线,提供足够的热量和营养素以改善营养状态。
避免烟雾和污染的空气等有害的环境暴露。
除了大剂量使用糖皮质激素和免疫抑制剂等需暂缓接种疫苗的情况以外,所有GDSD患儿应积极接种疫苗预防感染,尤其是流感疫苗、肺炎链球菌疫苗等。
合并急性感染时积极治疗,避免感染导致慢性肺病急性加重[44]。
GDSD的病理基础是肺间质慢性炎症,长期治疗主要使用抗炎药物。基于国内外个案或病例系列的经验,临床常用糖皮质激素、羟氯喹(Hydroxychloroquine)和小剂量大环内酯类药物。抗炎治疗主要用于SFTPB、SFTPC、ABCA3和NKX2.1变异引起的GDSD,其中对SFTPC变异的疗效相对较好;对CSF2RA、CSF2RB变异引起的PAP无效。
糖皮质激素是首选的抗炎药物,常用泼尼松1~2 mg/(kg·d)(或等效剂量甲泼尼龙)口服或静脉滴注,根据临床症状如呼吸、心率、SpO2改善情况,通常在治疗4~8周时评估治疗反应,也可能在数天内观察到呼吸和SpO2改善。伴严重低氧血症、呼吸衰竭威胁生命者,可采用大剂量糖皮质激素冲击疗法,甲泼尼龙10~30 mg/(kg·d),连续3 d,后续口服常规剂量泼尼松维持,1周左右初步评估疗效。糖皮质激素的疗程根据症状和肺部病变因人而异,控制病情后逐渐减量维持治疗,疗程可长达数年。短期大量应用糖皮质激素需注意消化道溃疡、消化道出血、高血压和高血糖等不良反应;长期应用可能诱发或加重感染、骨质疏松、股骨头坏死、生长发育障碍、青光眼、白内障和肾上腺皮质功能不全等。长期使用糖皮质激素注意同时补充维生素D和钙剂,定期监测生长曲线、眼部异常。
羟氯喹是4-氨基喹啉衍生物类抗疟药,有抗炎、调节免疫等作用,可能具有部分抗纤维化作用。国内外病例资料显示,对于糖皮质激素疗效欠佳的GDSD,尤其是SFTPC变异,羟氯喹可能有效[7,45,46]。糖皮质激素初始治疗失败者,可联合羟氯喹治疗,剂量为4~6 mg/(kg·d),6岁以上儿童最高可达10 mg/(kg·d)[47]。GDSD初始治疗也可以选择羟氯喹,尤其是不愿意使用糖皮质激素、不能耐受其不良反应或有禁忌证者。羟氯喹的潜在不良反应为视网膜色素沉着变化和视野缺损,但与药物剂量有很大相关性,在每日最大剂量不超过6.5 mg/kg情况下发生视网膜损害的风险低。建议在开始用药前进行眼部检查,用药后至少每年检查1次。
14、15元环的大环内酯类药物,如红霉素、克拉霉素、阿奇霉素、罗红霉素,有抗炎和调节免疫作用,推荐用于包括GDSD在内的各种ILD的治疗[44],临床上一般与其他抗炎药物联合治疗。常用红霉素5~10 mg/(kg·d),每日1次;或阿奇霉素5 mg/(kg·d),每日1次;或10 mg/(kg·d),每周3 d,疗程6个月至2年不等。大环内酯类药物的不良反应有消化道症状和肝毒性;少见心脏毒性,主要表现为QT间期延长和尖端扭转型室性心动过速,开始长期用药前可完善心电图等检查了解其诱发心脏毒性的可能性。小剂量应用不良反应较少见。
SP-B、SP-C、ABCA3或NKX2.1变异引起的严重GDSD,常规治疗无效者可进行肺移植。儿童肺移植目前仅在少数单位开展,截至2021年我国6个移植中心共开展了18例儿童肺移植手术[49],1例ABCA3变异成功接受肺移植,目前仍存活[15]。国外资料显示,GDSD患儿婴幼儿期肺移植后5年生存率为56%,儿童期肺移植后5年生存率为79%。肺移植远期并发症主要为高血压、移植后闭塞性细支气管炎、移植后淋巴组织增生症及肾功能不全等[50]。有报道1例CSF2RB变异的遗传性PAP在双肺移植后复发,其可能由骨髓来源的巨噬细胞介导[51],其他GDSD移植后无复发[50]。
基因治疗是GDSD最有前景的治疗方法,目前仍处于临床前研究阶段。基因治疗有不同的方法,如在体外、体内基因编辑(修正异常基因或使其失活),或体内导入外源性正常基因并表达,在细胞和动物实验水平均有成功的报道[54]。
GDSD需要长期治疗和管理。定期评估临床症状以及呼吸、心率、SpO2、肺功能、胸部CT以及心脏超声(监测肺动脉高压);监测生长发育情况,药物不良反应。可根据病情制定随访周期,或在治疗后1、2、3、6和12个月随访,病情稳定后可年度随访监测。
不同基因变异引起的GDSD,预后具有一定特征,又有个体差异。
SFTPB变异多表现为严重RDS,如不进行肺移植,绝大多数在3~6个月内死亡。
SFTPC变异具有可变外显率和表现度,即使携带相同的致病位点,不同个体的预后可能完全不同[7,55]。SFTPC变异主要表现为ILD,多数进展缓慢,可能存在稳定的慢性肺病,生存质量尚可;部分患者可能死亡或最终需要肺移植[56]。
ABCA3变异导致新生儿RDS,或儿童ILD,预后与ABCA3变异类型有关。功能完全丧失性变异导致严重RDS,多在出生后第1年内死亡或者需要肺移植;功能部分丧失的变异,多表现为暂时性新生儿RDS和儿童ILD,晚发型ILD的预后有个体差异[55],但5岁前的死亡率仍然相当高,部分患者最终也需要肺移植[4]。最近欧洲多中心研究表明,ABCA3变异存活超过1年的患儿中(平均年龄6.3岁),82%(36/44)没有肺移植依然存活,随着时间肺功能逐渐下降、胸部HRCT肺间质病变进展趋势[57]。
NKX2.1变异引起的新生儿RDS,经治疗多可缓解;引起的ILD存在不同程度的呼吸系统症状,大约1/3的病例需要平均18个月的吸氧治疗,部分死于呼吸衰竭或等待肺移植,成人患者可有肺纤维化[16,17]。
CSF2RA、CSF2RB变异引起的遗传性PAP常有重度的进展性肺病,全肺灌洗可能有效,但长期的疗效和预后仍有待观察。病例报告显示进行造血干细胞移植,植活后PAP好转[52,53]。
GDSD患儿检出的致病或可能致病的变异位点以及患儿父母该位点的基因测序结果,可为患儿家庭提供遗传咨询,如评估下一胎的患病风险,或辅助诊断家庭成员的疾病。
基因检测已成为诊断GDSD最重要的手段,但临床-影像-病理符合GDSD而基因检测未能找到致病基因的情况并不少见。除了对已知候选基因和变异位点的功能进行更深入研究,还要注意追踪和挖掘新的候选基因。新近报道RAB5B基因可调节表面活性物质的分泌通路,下调SP-B和SP-C表达,患者临床-影像-病理符合GDSD[58]。
基因诊断和致病位点的功能研究将为基因特异的个体化药物治疗奠定基础。针对ABCA3缺陷,新近报道通过对1 280种小分子进行筛选,发现环孢素A是某些变异位点特异性的ABCA3分子矫正剂,值得进一步临床研究[59]。
吡非尼酮和尼达尼布是2种常用于成人进展性纤维化性肺疾病的具有循证医学证据的抗纤维化药物,但目前在儿童还没有适应证。尼达尼布用于6~17岁儿童纤维化性肺疾病的随机安慰剂对照试验结果显示安全性良好[60],为有进展性肺纤维化的GDSD患儿应用这类抗纤维化药物奠定了基础。
基因治疗的临床前研究已取到较大进展。Alapati等[61]在小鼠子宫内应用CRISPR-Cas9介导SftpcI73T突变基因失活,使SP-C缺陷的小鼠胎儿和出生后小鼠的肺形态改善,存活率增加。Kang等[62]使用腺相关病毒6载体包装proSFTPB cDNA,经气管内给予使SP-B缺陷小鼠恢复表面活性物质的内稳态,防止肺损伤,改善肺生理,结果显著延长了SP-B缺陷小鼠的存活时间。
肺移植是挽救GDSD终末期肺病的有效措施,我国儿童肺移植有待积累更多临床经验[63]。
(陈杰华 姚瑶 刘秀云 申昆玲 执笔)
参与本共识制定和审校的专家(按姓氏拼音排序):陈杰华(深圳市儿童医院);陈星(山东第一医科大学附属省立医院);丁圣刚(安徽医科大学第一附属医院);蒋敏(广西医科大学第一附属医院);刘恩梅(国家儿童健康与疾病临床研究中心,重庆医科大学附属儿童医院);刘秀云(国家儿童医学中心,首都医科大学附属北京儿童医院,国家呼吸系统疾病临床医学研究中心);农光民(广西医科大学第一附属医院);沙莉(首都儿科研究所附属儿童医院);申昆玲(国家儿童医学中心,首都医科大学附属北京儿童医院,国家呼吸系统疾病临床医学研究中心,深圳市儿童医院);孙新(空军军医大学西京医院);唐素萍[福州市第一总医院儿童专科院区(福建省福州儿童医院)];姚瑶(国家儿童医学中心,首都医科大学附属北京儿童医院,国家呼吸系统疾病临床医学研究中心);张建华(上海交通大学医学院附属新华医院);郑跃杰(深圳市儿童医院);钟礼立(湖南省人民医院)
所有作者均声明不存在利益冲突

| 基因型 | 致病变异(Pathogenic) | 可能致病变异(Likely pathogenic) |
|---|---|---|
| SFTPB | c.1084-1G>A | c.904T>G (p.Cys302Gly)/C302G |
| c.883C>T (p.Arg295Ter)/R295* | c.730G>A (p.Gly244Ser)/G244S | |
| c.876G>A (p.Trp292Ter)/W292* | ||
| c.754C>T (p.Arg252Cys)/R252C | ||
| c.465G>T (p.Gly155=) | ||
| c.146G>A (p.Cys49Tyr)/C49Y | ||
| SFTPC | c.196G>A (p.Glu66Lys)/E66K | c.163C>T (p.Leu55Phe)/L55F |
| c.218T>C (p.Ile73Thr)/ I73T | c.192C>G (p.His64Gln)/H64Q | |
| c.329T>C (p.Leu110Pro)/L110P | c.314A>T (p.Asp105Val)/D105V | |
| c.347C>A (p.Ala116Asp)/A116D | c.316T>C (p.Tyr106His)/Y106H | |
| c.435+1G>T | c.334G>C (p.Ala112Pro)/A112P | |
| c.435+1G>A | c.337T>C (p.Tyr113His)/Y113H | |
| c.545T>A (p.Leu182Gln)/L182Q | c.397A>C (p.Ser133Arg)/S133R | |
| c.563T>C (p.Leu188Pro)/ L188P | c.435G>A (p.Gln145_Ala146=) | |
| c.435+2T>C | ||
| ABCA3 | c.4909+1G>A | c.4648C>T (p.Arg1550Trp)/R1550W |
| c.4772A>C (p.Gln1591Pro)/Q1591P | c.4618G>A (p.Glu1540Lys)/E1540K | |
| c.4658T>C (p.Leu1553Pro)/L1553P | c.4376G>A (p.Gly1459Asp)/G1459D | |
| c.4545C>G (p.Tyr1515Ter)/Y1515* | c.3905G>A (p.Gly1302Glu)/G1302E | |
| c.4195G>A (p.Val1399Met)/V1399M | c.3677T>C (p.Leu1226Pro)/L1226P | |
| c.3863-98C>T | c.3145T>C (p.Ser1049Pro)/S1049P | |
| c.3805G>T (p.Glu1269Ter)/E1269* | c.2883C>T (p.Gly961=) | |
| c.3426G>A (p.Trp1142Ter)/W1142* | c.2880G>C (p.Leu960Phe)/L960F | |
| c.3235C>T (p.Gln1079Ter)/Q1079* | c.2053-1G>C | |
| c.2880G>C (p.Leu960Phe)/L960F | c.629G>T (p.Gly210Val)/G210V | |
| c.2264-2A>G | c.622C>T (p.Arg208Trp)/R208W | |
| c.1939C>T (p.Gln647Ter)/Q647* | c.614-2A>G | |
| c.1702A>G (p.Asn568Asp)/N568D | c.557C>T (p.Pro186Leu)/P186L | |
| c.1455C>G (p.Tyr485Ter)/Y485* | c.440C>T (p.Pro147Leu)/P147L | |
| c.1455C>A (p.Tyr485Ter)/Y485* | c.233G>C (p.Trp78Ser)/W78S | |
| c.1444C>T (p.Gln482Ter)/Q482* | c.127C>T (p.Arg43Cys)/R43C | |
| c.1302G>A (p.Trp434Ter)/W434* | ||
| c.1286-2A>G | ||
| c.1240G>A (p.Ala414Thr)/A414T | ||
| c.743C>T (p.Pro248Leu)/P248L | ||
| c.302T>C (p.Leu101Pro)/L101P | ||
| c.128G>A (p.Arg43His)/R43H | ||
| NKX2.1 | c.1206A>C (p.Ter402Cys) | c.872C>G (p.Pro291Arg)/P291R |
| c.1066C>T (p.Gln356Ter)/Q356* | c.872C>T (p.Pro291Leu)/P291L | |
| c.872C>T (p.Pro291Leu)/P291L | c.728G>A (p.Arg243His)/R243H | |
| c.745C>T (p.Gln249Ter)/Q249* | c.727C>T (p.Arg243Cys)/R243C | |
| c.728G>T (p.Arg243Leu)/R243L | c.714G>A (p.Trp238Ter)/W238* | |
| c.727C>T (p.Arg243Cys)/R243C | c.713G>C (p.Trp238Ser)/W238S | |
| c.727C>A (p.Arg243Ser)/R243S | c.695C>T (p.Pro232Leu)/P232L | |
| c.714G>A (p.Trp238Ter)/W238* | c.664G>T (p.Glu222Ter)/E222* | |
| c.713G>T (p.Trp238Leu)/W238L | c.658G>A (p.Glu220Lys)/E220K | |
| c.703G>T (p.Val235Phe)/V235F | c.645C>A (p.Tyr215Ter)/Y215* | |
| c.695C>T (p.Pro232Leu)/P232L | c.617T>C (p.Leu206Pro)/L206P | |
| c.664G>T (p.Glu222Ter)/E222* | c.613G>T (p.Glu205Ter)/E205* | |
| c.645C>G (p.Tyr215Ter)/Y215* | c.583C>T (p.Arg195Trp)/R195W | |
| c.637C>T (p.Gln213Ter)/Q213* | c.436G>A (p.Ala146Thr)/A146T | |
| c.626G>C (p.Arg209Pro)/R209P | ||
| c.619G>T (p.Glu207Ter)/E207* | ||
| c.617T>A (p.Leu206Gln)/L206Q | ||
| c.613G>T (p.Glu205Ter)/E205* | ||
| c.612C>A (p.Tyr204Ter)/Y204* | ||
| c.596C>A (p.Ser199Ter)/S199* | ||
| c.524C>A (p.Ser175Ter)/S175* | ||
| c.464-2A>G | ||
| c.463+1G>A | ||
| c.432C>A (p.Tyr144Ter)/Y144* | ||
| c.391C>T (p.Gln131Ter)/Q131* | ||
| c.326C>A (p.Ser109Ter)/S109* | ||
| c.1A>T (p.Met1Leu)/M1L | ||
| CSF2RA | c.82C>T (p.Arg28Ter)/R28* | c.810+2T>A |
| c.414G>A (p.Trp138Ter)/W138* | ||
| c.586G>A (p.Gly196Arg)/G196R | ||
| c.595C>T (p.Arg199Ter)/R199* | ||
| c.610C>T (p.Gln204Ter)/Q204* | ||
| c.649C>T (p.Arg217Ter)/R217* | ||
| c.787C>T (p.Gln263Ter)/Q263* | ||
| c.917G>A (p.Trp306Ter)/W306* | ||
| CSF2RB | c.631del (p.Arg211fs) | 无 |
注:数据来源于Clinvar数据库(https://www.ncbi.nlm.nih.gov/clinvar),获取时间2022-12-13。致病和可能致病两者有重叠时为该位点的评级有不一致 Data from Clinvar database(https://www.ncbi.nlm.nih.gov/clinvar),access date 2022-12-13.The rating is inconsistent if there is an overlap of mutations between pathogenic and likely pathogenic group
























