
DNM1L基因变异所致线粒体和过氧化物酶体裂变缺陷相关脑病是一种罕见的、致死性的癫痫性脑病,具有临床表型和遗传异质性特点。急性期为药物难治性癫痫,预后差,可遗留严重神经系统后遗症。现报道1例经基因确诊的线粒体和过氧化物酶体裂变缺陷相关脑病患者,总结其临床资料及诊治过程,并进行文献复习,以提高对该疾病的认识。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
线粒体和过氧化物酶体裂变缺陷相关脑病(encephalopathy due to defective mitochondrial peroxisomal fission 1,EMPF1)是由DNM1L基因突变引起的罕见代谢病,导致在线粒体和过氧化物酶体裂变、融合过程中起重要调节作用的动力相关蛋白1(dynamin related protein 1,DRP1)出现缺陷,是一种致死性脑病,由Waterham等[1]于2007年首次描述。该病具有临床表型和遗传异质性特点,以常染色体显性遗传方式为主,少数可见常染色体隐性遗传。由于不同突变位点以及分裂缺陷程度对线粒体呼吸链和过氧化物酶体功能影响不同,EMPF1患儿发病年龄、特征和严重程度各不相同,从早发的严重致死性脑病伴或不伴癫痫发作,罕见的以“缓慢渐进性视力丧失”为主要表现的孤立视神经萎缩,到儿童期突然出现超级难治性癫痫持续状态,进而演变为全脑萎缩和严重致残脑病甚至死亡,预后极差。目前国外仅有 38例病例报道,国内报道病例相对较少,检索中文文献仅有4篇共6例病例报道[2, 3, 4, 5]。现将郑州大学附属儿童医院重症监护室确诊的1例EMPF1患儿临床特征和基因突变情况进行分析,发现的位点变异已被报道,但有新的临床表型,旨在提高临床医生对本病的认识,为临床诊断和遗传咨询提供依据。
临床资料 患儿女性,4岁9个月,因“双下肢抖动2 d余,抽搐伴意识障碍、机械通气1 d”于2022年9月13日入住郑州大学附属儿童医院重症监护室。入院2 d前患儿无明显诱因出现双下肢发软伴频繁不自主抖动,意识清楚,无发热、呕吐等伴随症状。1 d前患儿仍有频繁双下肢抖动,并渐转化为全身抖动、频繁抽搐发作,伴口唇发绀及经皮血氧饱和度下降,给予气管插管呼吸机辅助通气,院外给予“咪达唑仑(最大量15 μg· kg-1· min-1)、丙戊酸(1 mg· kg-1· h-1)及丙泊酚(1 mg· kg-1· h-1)”抗惊厥发作治疗,效果差。既往史:入院4个月前患儿无明显诱因出现双下肢肢体发软伴抖动情况,可自行缓解,意识清楚,无发热、咳嗽、呕吐及腹泻等伴随症状,完善脑电图检查未见异常,未行腰椎穿刺、头颅MRI检查。个人史及生长发育史:患儿系孕2产2、足月剖宫产,大运动及语言发育、智力水平正常。家族史:患儿母亲有乳腺癌病史。患儿堂哥有癫痫病史,未行基因检查。
体格检查:体温37.9 ℃,脉搏155次/min,呼吸25 次/min(机控),血压98/56 mmHg(1 mmHg=0.133 kPa),经皮血氧饱和度94%,体重14.5 kg,身高103 cm(-2倍标准差~-1 倍标准差),头围45. 4 cm(<-3倍标准差),尖下巴,昏迷,气管插管呼吸机辅助通气,双侧瞳孔等大等圆,直径2 mm,对光反射迟钝,心肺腹无异常,四肢肌张力阵发性增高,双侧膝、跟腱反射未引出,病理反射未引出,脑膜刺激征阴性。
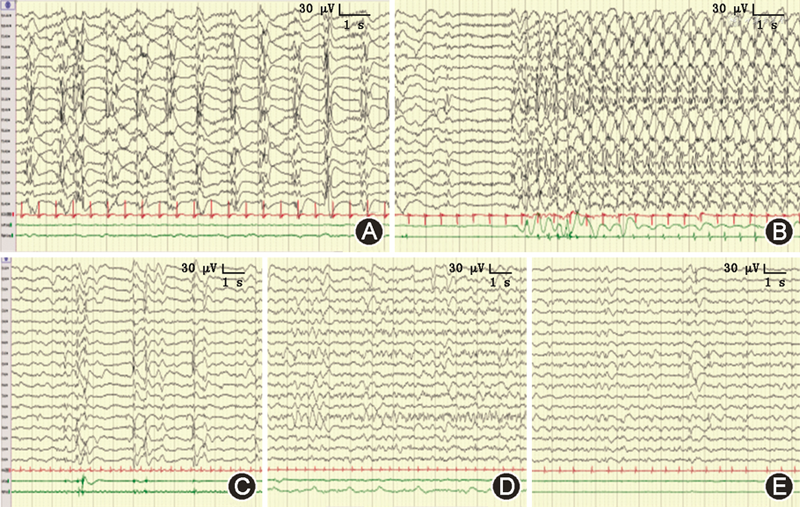
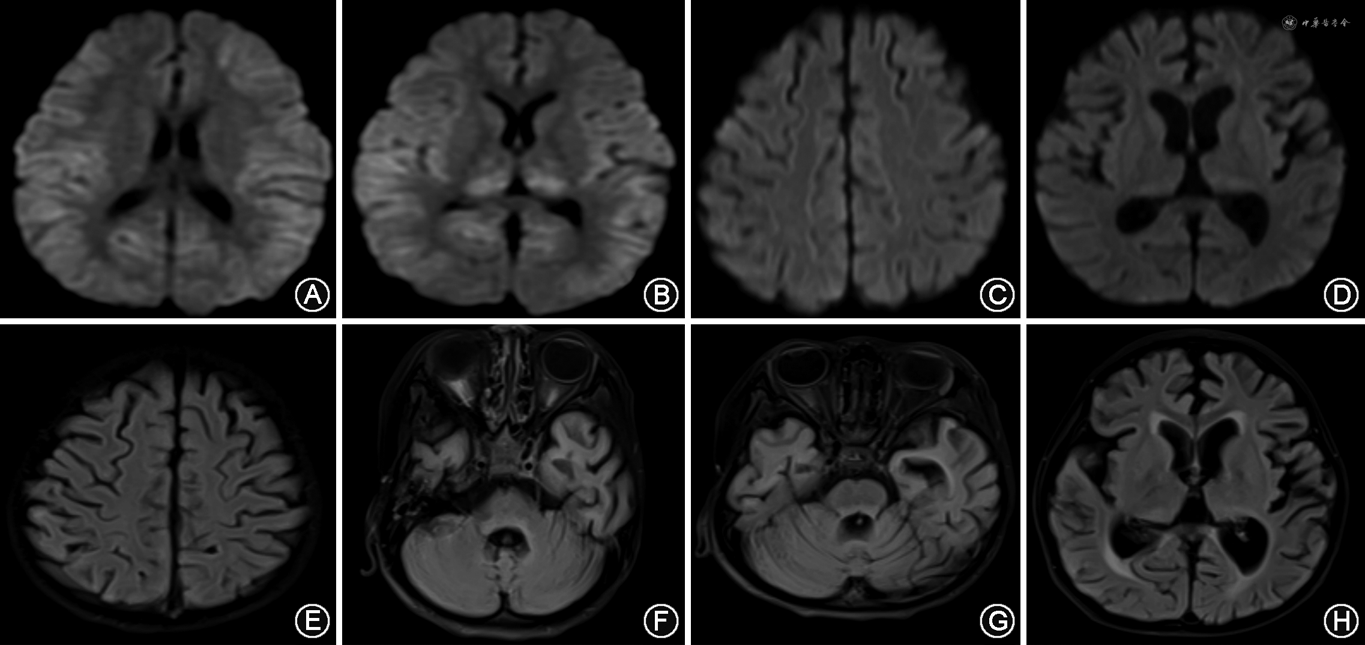
实验室及影像学检查:脑脊液常规、生化、涂片、培养及脑脊液宏基因组二代测序(metagenomic next-generation sequencing,mNGS)均正常,脑脊液乳酸2.87 mmol/L(正常值1.00~2.78 mmol/L),脑脊液白细胞介素6 17.66 pg/ml(正常值<5.30 pg/ml),血白细胞介素6 329.6 pg/ml;脑脊液及血液的自身免疫性脑炎相关抗体以及副肿瘤综合征抗体均阴性;血生化、血氨、血乳酸、铜蓝蛋白、同型半胱氨酸、丙酮酸、甲状腺功能均正常;血、尿遗传代谢未见异常;极长链脂肪酸水平正常;血毒物筛查未见异常。患儿视频脑电图动态改变见图1,头颅MRI动态改变见图2。
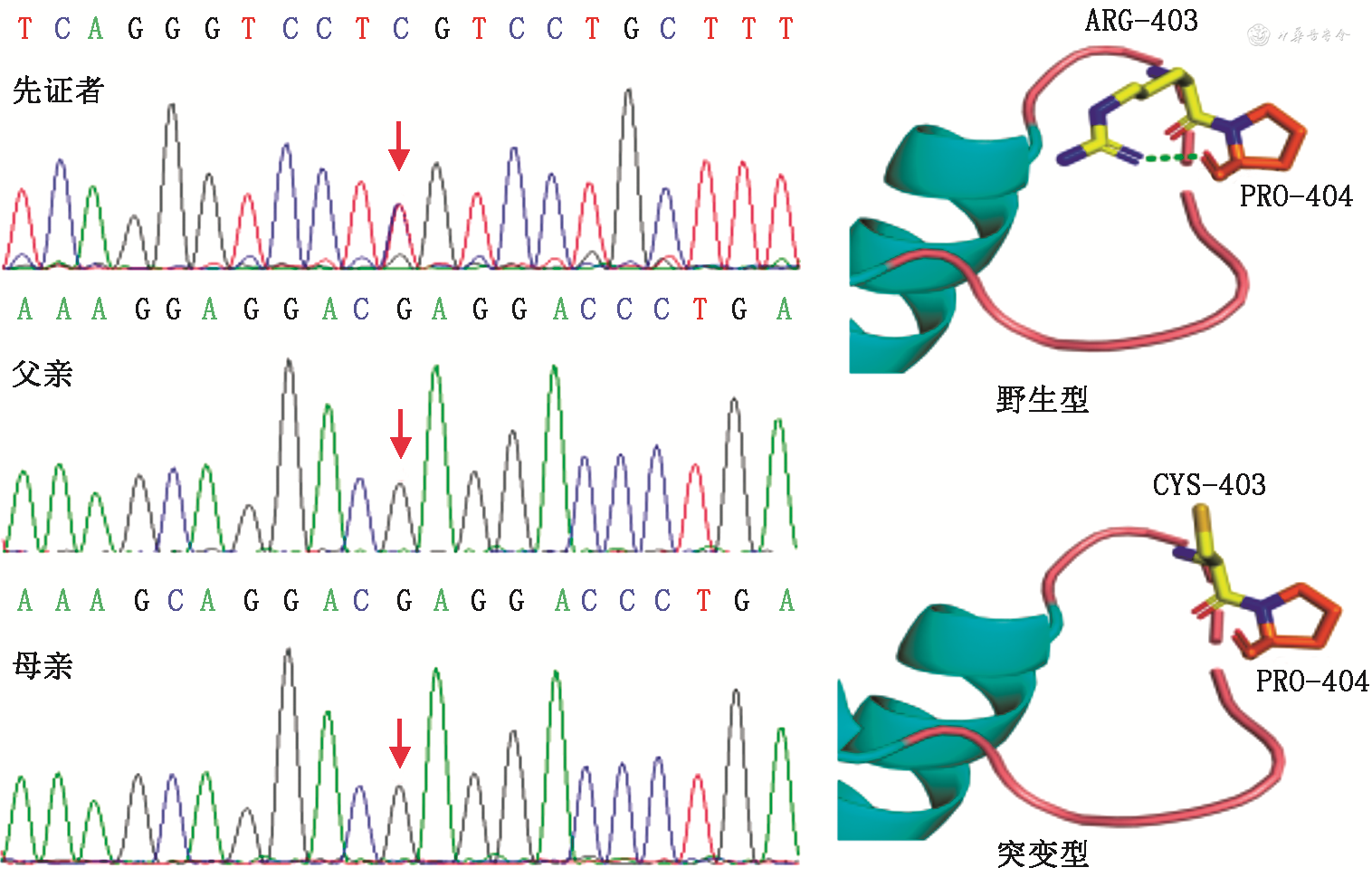
基因检测:在患儿家属签署知情同意书后,收集患儿及其父母外周血各2 ml,采用高通量测序平台进行全外显子组测序(委托北京迈基诺医学检验所完成),结果检测到DNM1L基因[NM_012062.5:c. 1207C>T(p. Arg403Cys)]位点杂合错义突变,即在1 207号核苷酸由胞嘧啶C变为胸腺嘧啶T,导致第403号氨基酸由精氨酸变为半胱氨酸。患儿父母基因为野生型,表型正常。我们用三维图分别表述上述野生型、突变型对蛋白质结构的影响(图3)。变异的生物信息学及致病性分析:生物信息学蛋白功能综合性预测软件 REVEL 预测结果为有害,SIFT、PolyPhen2、MutationTaster、GERP+预测结果均为有害(PP3);在正常人群数据库中的频率为0(PM2);文献数据库已有该位点(encephalopathy,infantile)的病例报道,变异标签为 DM(致病突变);ClinVar 数据库对该位点的致病性分析结果为:致病性的视神经萎缩症5型|脑病|有害|线粒体过氧化物酶体裂变1缺陷|先天性遗传性疾病|未提供(PS4);经家系验证分析,受检人之父该位点无变异,受检人之母该位点无变异,此变异为自发突变(PS2),根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)指南评级为致病性(PS4+PS2+PM2+PP3)。
治疗经过及随访:患儿入院时为已超过24 h的超级难治性癫痫持续状态,给予咪达唑仑(最大剂量30 μg·kg-1·min-1),丙戊酸钠(最大剂量40 mg·kg-1·d-1),加用左乙拉西坦(最大剂量60 mg·kg-1·d-1)、苯巴比妥(5 mg·kg-1·d-1)及盐酸艾司氯胺酮(最大剂量4.3 mg·kg-1·h-1)抗癫痫发作,同时给予经验性抗感染,甲泼尼龙冲击(20 mg·kg-1·d-1,共3 d)、丙种球蛋白(1 g·kg-1·d-1,共2 d)调节免疫等治疗,患儿脑电图持续呈现广泛性周期性痫样放电。
入院第6天启动全肠内生酮饮食,多次监测脑电图,抽搐发作仍不能控制,并转化为肌阵挛持续状态。结合患儿脑脊液白细胞介素6升高,入院第13天、第27天分别给予托珠单抗免疫治疗(每次4 mg/kg),患儿抽搐发作频繁,期间先后给予加用拉考沙胺(最大剂量10 mg· kg-1· d-1)、托吡酯(最大剂量10 mg· kg-1· d-1,应用3 d后因皮疹停用)、氯硝西泮(最大剂量0.2 mg· kg-1· d-1)、唑尼沙胺(最大剂量8.3 mg· kg-1· d-1)等多种药物,病程中因严重肝功能损害,先后停用丙戊酸、苯巴比妥。入院后3周左右给予拔除气管插管。入院第6周基因测序结果回报,明确诊断为“EMPF1”,给予线粒体鸡尾酒疗法,并加用吡仑帕奈(最大剂量0.12 mg· kg-1· d-1)抗癫痫治疗,氯巴占(最大剂量0.83 mg· kg-1· d-1)逐渐替代氯硝西泮,患儿四肢肌张力高,联合盐酸苯海索(最大剂量0.25 mg· kg-1· d-1)缓解肌张力。
入院后2个月患儿无临床发作,脑电图提示弥漫性慢波活动、阵发性痫样波发放,最终抗发作药物调整为:拉考沙胺(10 mg· kg-1· d-1)、唑尼沙胺(8.3 mg· kg-1· d-1)、氯巴占(0.83 mg· kg-1· d-1)、吡仑帕奈(0.12 mg· kg-1· d-1)及苯海索(0.25 mg· kg-1· d-1)联合应用,继续生酮饮食、鸡尾酒疗法。头颅MRI提示进行性脑萎缩,呈持续昏迷状态,共住院2个月余出院。末次随访为病史8个月(2023年5月),患儿意识好转,可经口进食,但遗留严重认知功能障碍,癫痫发作5~6次/d,表现为刺激后出现不自主肢体抖动,每次持续数秒钟自行缓解。
DNM1L基因位于第12号染色体,包含19个外显子,其编码的DRP1具有736个氨基酸,包含3个动力蛋白样鸟苷三磷酸酶(guanosine triphosphatase,GTPase)类的典型结构域,分别为N端GTPase头部结构域,中间结构域(MD)和C端GTPase效应结构域(GED)。DRP1在细胞质中产生,借助外膜上的受体募集到线粒体表面,组装成低聚物环驱动线粒体小管的收缩和断裂,所有影响上述结构域的致病变异最终会影响DRP1的寡聚化和自组装[6]。由于DRP1在脑组织中高表达,控制大脑发育和神经元活动,因此,线粒体以及过氧化物酶体裂变缺陷可引起复杂的神经系统表型,在多数研究中被描述为一种致死性脑病。肌张力障碍、痉挛发作、小头畸形、疼痛不敏感、周围神经病变、视神经萎缩和意识障碍等是DNM1L变异相关的常见临床特征[1,7, 8, 9, 10, 11, 12, 13, 14, 15]。
复习文献发现,c.1207C>T(p.R 403C)为DNM1L热点突变基因,大多数该位点突变者表现为类似的临床病程,患者似乎都经历了一段基本正常的发育时期,但在儿童时期出现轻微代谢损伤(疫苗接种、呼吸道感染、颅脑外伤)后发展为癫痫持续状态。感染、免疫和血代谢评估大多为阴性,肌肉活组织检查显示组织学和线粒体呼吸链酶多数也正常,MRI特征性表现为进行性脑萎缩,累及海马及丘脑。病情进展相似,包括难治性癫痫、严重脑病、发育退化、肌阵挛等,一旦癫痫发作,局灶性或全身性癫痫持续状态的反复发作往往会引发认知退化,常见儿童早期死亡[9, 10, 11, 12, 13]。本例患儿全外显子组测序显示是新发变异(c.1207C>T,p.R 403C),影响DRP1中间结构域中的一个关键氨基酸残基,即第403号氨基酸由精氨酸变为半胱氨酸。结合既往文献报道,本例患儿具有突出的临床表型,即该患儿半年内出现2次发作:第1次无诱因出现局灶运动性发作,自行缓解,未出现认知功能障碍;间隔数月后再次出现局灶运动性发作并进展为全面强直-阵挛发作。且患儿处于持续昏迷状态,遗留严重神经系统后遗症。与本病例相似,既往仅Minghetti等[16]报道DNM1L脑病可表现为双相临床进展:先证者6岁时出现第一次肌阵挛发作,应用抗发作药物后抽搐得到控制,并重新融入社会和学校,间隔13年后出现第二次发作并呈癫痫持续状态,文章发表时患者已为持续性植物状态。因此,对DNM1L脑病患者需进行长期随访,患者可能表现出明显延长的缓解期,癫痫控制满意甚至无需药物控制,但其潜在的线粒体病因最终会导致患儿毁灭性的全脑萎缩及极差的神经系统预后。
DNM1L突变表现出广泛的表型谱和不同的严重程度,从早期死亡到缓慢进展性脑病伴肌张力减退、发育迟缓,伴或不伴癫痫发作,多数为DNM1L常染色体显性遗传,且为新发突变。文献报道新发杂合变异c.1184C>A(p.A395D)先证者因严重神经功能障碍,生后37 d死亡[1]。Assia Batzir等[17]和Lhuissier等[18]报道的新发杂合变异患者,虽有发育迟缓、肌痉挛发作等严重脑病表现,但可存活到成年。而Sheffer等[14]报道的新发杂合c.1084G>A(p.G362S)突变者则表现为慢性神经系统障碍,以产后小头畸形、发育迟缓和疼痛不敏感为特征,临床表现相对较轻。DNM1L变异可表现为隐性遗传,但严重程度亦不相同[19, 20, 21]。因此,受影响的个体有不同表型,其发病年龄、症状和严重程度有显著差异,一些患者甚至没有典型的临床/生化症状。考虑可能是由于变异类型和位置不同,对结构域功能影响不同。既往研究结果表明,基因内突变的定位决定了对DRP1寡聚化和随后的线粒体裂变活性的显性负影响程度,可导致明显不同程度的脑病和临床结局:早期死亡和长期癫痫性脑病[22, 23]。动物模型研究结果也显示构成纯合子的DNM1L基因敲除,小鼠在胚胎发育过程中即发生死亡,而小鼠大脑中有前提条件的DNM1L敲除则导致发育缺陷,这两种情况都与线粒体裂变受损有关[24, 25]。其次,对线粒体相关功能的评估结果也表明,致病性变异基因与临床意义未明的变异共存,后者可作为修饰剂影响疾病表型。
DNM1L突变主要影响神经系统,还可累及骨骼肌、心肌,尸检发现骨骼肌及心肌细胞均呈肌病特征[26]。小鼠模型研究也证明DNM1L基因突变可导致常染色体显性扩张性心肌病并最终发生充血性心力衰竭[27, 28]。因此,对于DNM1L突变患者需监测心脏超声,进行心功能动态评价。此外,Piccoli等[23]报道了1例DNM1L变异患者,该先证者母亲分别与不同男子连续生育了2例具有相同表型的癫痫性脑病患儿,基因测序排除了X-连锁,考虑先证者母亲为生殖腺嵌合体。因此,对于以新发变异为主要变异类型的DNM1L基因,即使外周血基因检测结果显示先证者母亲未携带致病变异,再次妊娠时仍建议进行产前基因诊断。
目前对DNM1L脑病无有效根治方法,治疗主要是对症及支持治疗。根据临床表现和脑电图发作形式,排除结构/代谢/感染因素后,患者可能被误诊为热性感染相关性癫痫综合征、自身免疫性脑炎等,给予大剂量咪达唑仑、氯胺酮、丙泊酚等麻醉药以及丙戊酸、左乙拉西坦、托吡酯等多种抗发作药物治疗无效。经验性给予大剂量激素、免疫球蛋白和血浆置换等一线免疫调节治疗,以及阿纳白细胞介素、利妥昔单抗等二线免疫调节药物均无效。急性期呈现药物难治性癫痫发作,文献报道似乎只有巴比妥酸盐昏迷疗法可导致脑电图爆发抑制和临床癫痫发作缓解。生酮饮食也没有明确获益,但部分患者是因生酮饮食不良反应而终止使用[10, 11, 12,15, 16]。本例患儿在多种抗发作药物治疗失败的基础上加用生酮饮食,且无明显不良反应,最终癫痫发作得到控制,生酮饮食作为辅助治疗,似乎有一定疗效及安全性,但缺乏大样本量研究及随机对照试验证实。本研究及Minghetti等[16]报道的患者呈现双相病程,后期均出现严重癫痫脑病,由于潜在的线粒体病因,因此,及时治疗也不一定能避免临床进展,该病目前尚无特效药物预防。此外,由于癫痫持续状态发作,患者可能需要在重症监护室中长期滞留,合并严重感染时可加重疾病表型。Mancardi等[10]和Nolan等[12]报道的患儿均因前期大剂量使用免疫抑制剂,病程中反复发生肺部和尿路感染,最终均死于严重脓毒血症。因此,治疗过程中需注意控制感染,病情允许情况下尽早撤离呼吸机、减少呼吸机相关肺炎发生。本例患者入院后3周左右撤离呼吸机,无严重院内感染发生,可能是较其他突变者预后相对好的一项因素。
我们报道了先证者DNM1L的新发错义突变,并将这种突变的临床特征与既往文献报道进行比较,患者病程中表现为双相临床进展,扩展了DNM1L相关线粒体裂变缺陷的表型。DNM1L脑病为药物难治性癫痫,尽早启动生酮饮食以及治疗过程中注意感染可能有助于控制癫痫。同时,对于那些既往生长发育正常、发病年龄晚的难治性癫痫脑病患者,特别是临床快速恶化的患者,均建议尽早进行全外显子组测序。
杜语慧, 贾鑫磊, 梅道启, 等. DNM1L基因变异致线粒体和过氧化物酶体裂变缺陷相关脑病 1例[J]. 中华神经科杂志, 2024, 57(1): 74-79. DOI: 10.3760/cma.j.cn113694-20231011-00227.
所有作者声明无利益冲突
None declared

























