
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患者男,59岁。2008年无明显诱因出现腹痛、腹胀,以右腹及脐周明显;2011年12月突发性小肠穿孔,弥漫性腹膜炎,外院行小肠穿孔修补术,术中见"小肠、结肠多发憩室"。术后切口经3个月愈合,并形成腹壁切口疝。术后腹胀、腹痛、腹泻未缓解。2012年9月,腹痛、腹胀症状加重,立位腹平片示大量肠管积气扩张,以左上腹著,考虑为"小肠不全梗阻",在我院行剖腹探查术。术中见Treitz韧带以下小肠及结肠多发憩室,以近端小肠为甚,行近端小肠部分切除术及切口疝修补术。术后切口经4个月愈合。术后给予美沙拉秦、双歧杆菌三联活菌对症治疗,腹痛、腹胀、腹泻症状明显好转。2013年10月,胃镜示胃扭转,胃窦变形,十二指肠球部溃疡。胸部CT示双上肺气肿;轻度肺动脉高压[43 mmHg(1 mmHg=0.133 kPa)]。十二指肠降段、结肠、膀胱后壁多发憩室。入院后继续给予埃索美拉唑镁、硫糖铝治疗十二指肠溃疡,症状减轻。
病理检查:大体检查:切除小肠肠管一段,长51 cm,周径5.0~7.5 cm,距一断端22 cm、另一断端27 cm处可见管腔狭窄,狭窄区肠腔直径约2 cm,部分黏膜皱襞粗大,并见多个息肉样隆起,直径0.2~0.8 cm,肠系膜侧见多个憩室,直径约0.2~1.3 cm,肠壁未见明显增厚,靠近狭窄处肠黏膜水肿,水肿黏膜面积5.5 cm×7.0 cm,余黏膜光滑。光镜观察:肠黏膜尚完整,黏膜及黏膜下层不规则隆起并突入肠腔(图1,图2),黏膜固有层慢性炎性细胞浸润,绒毛及隐窝结构规则,黏膜下层血管增多,走形迂曲,管腔不规则,管壁厚度尚均一,局灶见新鲜及陈旧性出血(图3,图4);隆起处黏膜下层见多个大小不等薄壁囊腔,囊腔互不交通,囊腔形态不规则,偶见于浆膜下层,直径最大达0.8 cm,囊壁由纤维组织包绕而成,囊壁腔缘侧可见较多组织细胞及多核巨细胞,未见上皮衬覆(图5,图6);囊腔与迂曲的血管交叉分布;多灶环形肌层中断,纵行肌层尚存,环形肌缺失处黏膜及黏膜下层疝入肠周脂肪组织,被覆薄层纵行肌或纤维组织,形成憩室状结构。免疫组织化学染色:不规则囊腔衬覆细胞CD68染色阳性,CD34、D2-40染色阴性,证实为组织细胞。
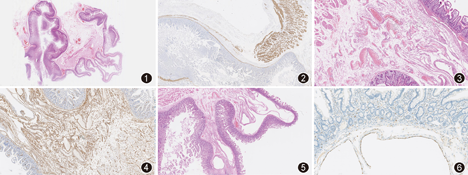
本例送检小肠标本在缺乏临床相关病史的情况下病理诊断为"小肠多发性假性憩室"。在临床病理讨论中进一步发现患者双手指关节、腕关节活动度大,关节易脱位,轻微损伤后易出现皮肤瘀斑、血肿,全身皮肤松弛,可向外拉伸4~5 cm;妹妹有类似症状,因肠穿孔手术后去世。经与临床共同讨论后发现,患者临床表现以消化道症状为主,小肠、结肠、膀胱多发性憩室,伴小肠穿孔,术后伤口长期不愈合;关节活动度大,全身皮肤松弛;并有家族史,最终明确诊断。
病理诊断:血管型Ehlers-Danlos综合征累及小肠,伴肠道多发憩室及肠气囊肿症。
讨论:Ehlers-Danlos综合征(EDS)是一种遗传性结缔组织病,是一组累及多系统、具有临床异质性的疾病,主要累及皮肤、韧带、关节、血管和内脏。目前认为,多数病例是由于编码胶原纤维或胶原合成酶基因突变所导致,并涉及其他细胞外基质的生物合成、信号转导及分子间相互作用[1]。传统上,根据基因缺陷、遗传方式及临床表型将EDS分为6型:经典型、高度可动型、血管型、脊柱后侧凸型、关节松弛型和皮肤伸展型,其中经典型、高度可动型和血管型最多见[2]。EDS共同临床特征是皮肤脆弱易损伤,皮肤延展性显著增加和关节过度活动。
血管型EDS发病率为1/100万~1/10万,是Ⅲ型胶原(COL3A1)胚系基因杂合性突变导致,由于Ⅲ型胶原大多分布于血管壁、空腔脏器,因此常见并发症包括血管破裂、肠道穿孔、妊娠子宫破裂等,病死率高达25%~50%,是所有EDS中预后最差的一型。其他临床特征包括皮肤薄、血管易见、易挫伤、具有特征性面容,但皮肤延展性过度和关节过度活动不如其他几型明显,临床上常被忽略,多数患者在出现并发症后才得到诊断。患者在儿童期的并发症罕见,20岁时25%的患者出现并发症,到40岁时这一比例高达80%。致命原因主要是动脉破裂,25%的病例累及消化道,出现消化道穿孔,需要行肠管切除[3]。患者肠管大体和光镜下均可见多灶性固有肌层变薄甚至缺失,常伴多发性憩室形成,一般无明显的血管异常[4],可能被误诊为憩室穿孔,此时要与其他原因导致的憩室病鉴别。对于血管型EDS,肠壁黏膜下层和固有肌层Ⅲ型胶原的减少高度提示该病的可能,生化检查证明Ⅲ型胶原质或量的缺陷可确诊95%的EDS,但COL3A1无义突变电泳检查可正常,因此DNA检测是诊断EDS的金标准。但患者小肠多发憩室,黏膜下层多见气囊肿形成,伴血管畸形,同时临床上关节活动度大、皮肤松弛,并有明确的家族史,综合考虑EDS诊断比较明确。
憩室在西方多见,可能与遗传背景、饮食习惯(低纤维素饮食)以及缺乏运动有关。憩室形成的主要因素为肠壁局部薄弱,此外肠腔压力增加(如肠道功能紊乱致肠道收缩频率增加、神经肌肉疾病等)也是憩室诱发和加重的重要因素。肠壁滋养血管穿越环形肌层处是局灶肠壁薄弱的解剖学因素,多数憩室于此处形成,并与憩室的并发症——出血相关。不同部位的憩室发病原因不尽相同:左半结肠,特别是乙状结肠,主要为年龄相关性憩室,西方多见,随着年龄增加,肠壁胶原分子结构和分布发生改变,为假性憩室(仅包含黏膜及黏膜下层,环形肌层缺如),一般为多发性,无黏膜下异常及肠道神经丛异常。右半结肠,多为先天性憩室,亚洲多见,为真性憩室(憩室壁含正常肠管的4层结构),单发或少发性(一般少于15个),部分病例伴血管结构不良(angiodysplasia)或先天性血管畸形[5]。与遗传性胶原弹力纤维异常相关的综合征有关,如Ehler-Danlos、Marfan、Williams-Beuren综合征等,憩室不仅累及结肠,还常累及小肠及其他空腔器官(如子宫),并伴血管病变等;此外,肿瘤由于可分泌多种基质金属蛋白酶,降解细胞外基质,可发生肿瘤相关性憩室,一般无症状。上述因素均应列入肠道多发性憩室、不明原因穿孔的鉴别诊断。对于小肠、结肠多发性憩室,伴或不伴其他空腔器官憩室(如膀胱)或破裂(如妊娠子宫),则需要考虑到相关综合征的可能。充分结合临床相关病史及体征是正确诊断的关键。
有关血管型EDS肠道病理改变的报道很少。肠壁Ⅲ型胶原减少导致肠壁弹性和强度降低,肠壁薄弱,是形成肠道多发憩室、肠穿孔的重要解剖学基础。大体上以固有肌层萎缩,显著变薄、甚至缺失,肌层薄弱区憩室形成,伴纤维化为特点。镜下见多灶肌层缺失,伴或不伴憩室形成、血管异常,黏膜下扁平,可见局灶致密的纤维组织。免疫组织化学Ⅲ型胶原染色可显示憩室处黏膜下层及固有肌层Ⅲ型胶原显著减少,在EDS患者相对正常的肠管,Ⅲ型胶原减少仅见于黏膜下层[4]。
本例另一个重要的病理学改变是肠壁黏膜下及浆膜下层肠气囊肿的形成。肠气囊肿(pneumatosisintestinalis)是指肠壁内和肠系膜出现气体,伴或不伴腹腔内游离气体,空肠最常累及,但胃及结直肠亦能发生,病变弥漫分布或仅累及一段或数段不相连肠管。肠气囊肿症的成因并不十分清楚,可能的理论包括:(1)机械性理论:肠梗阻、炎症性肠病、缺血性肠病、消化道肿瘤、肛门直肠手术、肠道准备或结肠镜等,导致肠壁损伤或肠腔内压力增加,可能是导致肠壁内积气的原因,但这一理论不足以解释积气在肠壁内如何形成并维持;(2)肺源性理论:肺部疾病,如慢性阻塞性肺疾病、哮喘、间质性肺炎等,可导致肺泡破裂,形成纵隔气肿,继而沿主动脉、肠系膜血管,到达肠壁;(3)细菌理论:肠壁内憩室是由于产气细菌通过黏膜屏障在肠壁内种植,间接证据是抗生素治疗肠气囊肿症有效,但囊肿内产气细菌无直接证据证实,并且囊肿破裂的患者无腹膜炎症状,进一步支持囊肿内气体不是细菌导致;(4)化学理论或营养不良理论:营养不良可影响碳水化合物的消化,增加肠道内细菌发酵,导致肠腔内大量气体,使得肠腔扩张、缺血,继而导致黏膜下气体囊肿形成。支持性证据是用α-葡糖苷酶抑制剂治疗肠气囊肿症有效[6]。多数情况下肠气囊肿症不引起症状,常为X线检查、破腹探查甚至尸检时偶然发现,偶尔黏膜下层气肿表面黏膜可糜烂出血。但在结缔组织病,如干燥综合征、皮肌炎、系统性红斑狼疮等,肠气囊肿可导致严重的急腹症而致命[7]。本例病变中,肠黏膜下层的肠气囊肿导致黏膜息肉样隆起、黏膜肿胀,肠腔狭窄,可能与患者腹痛、腹胀相关,并可能是导致该患者"小肠不完全梗阻"的因素之一。此外,肠气囊肿可导致肠腔压力增加,进一步加重肠憩室、肠穿孔。结缔组织病发生肠气囊肿的可能机制包括肠道肌层萎缩和纤维化导致肠道蠕动受损,从而细菌过生长,肠腔压力增加,并且肠壁渗透性增加等。因此,对于这类患者,调节肠道菌群和抗生素的使用可能能够起到缓解症状的作用。

























