世界卫生组织(WHO)"造血和淋巴系统肿瘤分类(2008年版)"中,将"嗜酸性粒细胞增多及血小板衍生的生长因子受体(PDGFR)α,β或成纤维生长因子受体(FGFR)1异常的髓系、淋巴系肿瘤(MLN-eo)"作为一类疾病单独列出。2011年,维也纳医学院组织的由多学科专家参与的"嗜酸性粒细胞相关疾病工作会议"结合既往诊断标准,就嗜酸性粒细胞增多相关疾病的诊断和分类进行了更新,但迄今为止,嗜酸性粒细胞增多相关疾病的分类、命名仍然比较混乱。笔者拟就近年嗜酸性粒细胞增多相关疾病文献进行复习,对嗜酸性粒细胞增多相关疾病,特别是克隆性嗜酸性粒细胞增多相关疾病的分类和诊疗进展进行综述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Hardy与Anderson[1]于1968年首次报道高嗜酸性粒细胞综合征(hypereosinophilic syndrome,HES),Chusid等[2]于1975年提出HES的具体诊断标准。近年来,随着分子生物学、免疫学技术在HES中的广泛应用,人们对嗜酸性粒细胞增多相关疾病的认识不断深入,对其进行了更加细化的分类,其诊治方案也随之不断发生变化。笔者拟就嗜酸性粒细胞增多相关疾病的分类及诊疗进展进行综述,旨在提高对嗜酸性粒细胞增多相关疾病的认识,从而正确选择及时有效的治疗措施,以改善患者预后。
正常个体外周血中嗜酸性粒细胞计数为(0.05~0.50)×109/L,当机体嗜酸性粒细胞计数≥1.5×109/L,且持续1个月以上,即为嗜酸性粒细胞增多[3]。根据发病原因的不同,嗜酸性粒细胞增多可以分为继发性、特发性及克隆性嗜酸性粒细胞增多。
继发性嗜酸性粒细胞增多主要继发于寄生虫感染、变态反应、血管炎及淋巴瘤等疾病。经染色体核型分析、分子生物学检测发现,此类增多的嗜酸性粒细胞均无核型及基因异常,不伴骨髓和外周血中原始细胞增多[4]。但Tabata等[5]和Tefferi[6]认为,伴有克隆性T淋巴细胞异常的嗜酸性粒细胞增多,曾被称为淋巴细胞变异型HES(lymphocytic variant HES),也应纳入为继发性嗜酸性粒细胞增多性疾病[5,6]。
特发性嗜酸性粒细胞增多是一类除继发性和克隆性嗜酸性粒细胞增多外的嗜酸性粒细胞增多性疾病[4],主要包括特发性嗜酸性粒细胞增多综合征(idiopathic hypereosinophilic syndrome,IHES)和特发性嗜酸性粒细胞增多症(idiopathic hypereosinophilia,IHE)。IHES诊断标准如下:①嗜酸性粒细胞增多(嗜酸性粒细胞计数≥1.5×109/L)持续6个月以上;②无引起嗜酸性粒细胞增多的潜在原因,如寄生虫感染、变态反应等;③嗜酸性粒细胞可浸润器官及引起功能障碍;④无克隆性嗜酸性粒细胞的证据;⑤无免疫表型异常[6]。而IHE是指缺乏上述条件③的一类特发性嗜酸性粒细胞增多性疾病[4]。需要注意的是,特发性嗜酸性粒细胞增多性疾病可能包含一些实际上为嗜酸性粒细胞白血病,或一些不明原因引起的继发性嗜酸性粒细胞增多性疾病,只是按照目前的技术条件难以鉴别诊断,未能进一步加以区分[7]。
在世界卫生组织(World Health Organization,WHO) "造血和淋巴组织肿瘤分类(2008年版)"中指出,克隆性嗜酸性粒细胞增多是指经组织学、细胞遗传学、分子学检查发现克隆性嗜酸性粒细胞的证据 [8]。克隆性嗜酸性粒细胞增多主要包括2大类:①慢性嗜酸性粒细胞白血病非特指型(chronic eosinophilic leukemia not otherwise specified,CEL-NOS);②伴嗜酸性粒细胞增多及血小板衍生的生长因子受体(platelet-derived growth factor receptor,PDGFR)α,β或成纤维细胞生长因子受体(fibroblast growth factor receptor,FGFR)1异常的髓系、淋巴系肿瘤(myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRα,PDGFRβ or FGFR1,MLN-eo)[9]。
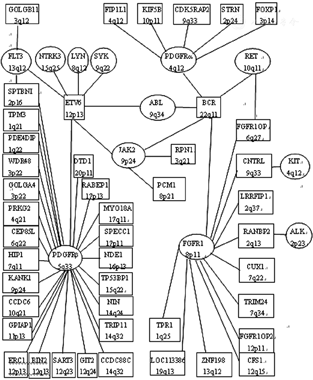
克隆性嗜酸性粒细胞增多通常与骨髓增殖性肿瘤(myeloproliferative neoplasm,MPN)相关[10]。目前,已经发现50多种不同融合基因与克隆性嗜酸性粒细胞增多相关疾病有关(图1)。细胞遗传学分析发现,克隆性嗜酸性粒细胞增多相关疾病中较为常见的基因断裂点可分为4种:位于4q12的PDGFRα,位于5q31-33的PDGFRβ,位于8p11-12的FGFR1及位于9p24的Janus激酶(Janus kinase,JAK)2[11]。
伴嗜酸粒细胞增多及FIP1L1-PDGFRα融合基因呈阳性的髓系、淋巴系肿瘤诊断标准:①MPN伴嗜酸性粒细胞增多;②FP1L1-PDGFRα融合基因呈阳性。伴嗜酸性粒细胞增多及FIP1L1-PDGFRα融合基因呈阳性的急性髓细胞白血病(acute myelocytic leukemia,AML)或淋巴细胞母细胞白血病/淋巴瘤也归于此类疾病。如果MPN患者出现CEL-NOS的血液学特征且Ph染色体呈阴性,伴脾大及血清维生素B12水平明显升高、血清类胰蛋白酶水平升高、骨髓肥大细胞增多,虽无法进行分子学分析,也应对患者进行伴嗜酸粒细胞增多及FIP1L1-PDGFRα融合基因呈阳性的髓系、淋巴系肿瘤的鉴别诊断。
伴ETV6-PDGFRβ或其他涉及PDGFRβ基因重排的MLN-eo诊断标准:①MPN伴嗜酸性粒细胞增多,有时伴中性粒细胞或单核细胞增多;②出现t(5;12)(q31-q33;p12)或涉及PDGFRβ的其他染色体易位,或ETV6-PDGFRβ融合基因呈阳性。由于t(5;12)(q31-q33;p12)并非均会导致ETV6-PDGFRβ融合基因呈阳性,因此应进行相关分子学分析鉴定。如患者表现为嗜酸性粒细胞增多、存在5q31-33断裂点易位且Ph染色体呈阴性的MPN,虽无法进行分子学分析,也应当对患者进行伴ETV6-PDGFRβ或其他涉及PDGFRβ基因重排的髓系、淋巴系肿瘤的鉴别诊断。
伴FGFR1重排的MPN或急性白血病的诊断标准:①MPN或AML、前体T/B淋巴母细胞白血病/淋巴瘤伴嗜酸性粒细胞明显增多;②因t(8;13)(p11;q12)或其他染色体易位导致髓系细胞或(和)原始淋巴细胞出现FGFR1重排。
根据上述诊断标准,不难发现:①WHO并未严格区分MLN-eo与伴嗜酸性粒细胞增多的MPN或伴嗜酸性粒细胞增多的MPN/骨髓增生异常综合征(myelodysplastic syndrome,MDS);②涉及PDGFRβ基因重排的疾病与髓系肿瘤关系密切;③PDGFRα的伙伴基因(基因突变后的配对基因)常为FIP1L1,FIP1L1-PDGFRα融合基因呈阳性的患者具有相对特异的临床表现;④PDGFRβ的伙伴基因较多,并非一定为ETV6(图1)。
伴嗜酸性粒细胞增多及PDGFRα基因异常的髓系、淋巴系肿瘤的临床表现:FIP1L1-PDGFRα融合基因呈阳性者常为CEL-NOS,AML或T淋巴母细胞淋巴瘤(T lymphoblastic lymphoma,T-LBL)伴嗜酸性粒细胞增多[12],主要临床表现为乏力、皮肤瘙痒,大部分患者脾大,少部分患者肝大。由于肺纤维化,患者可能出现呼吸困难、咳嗽。最严重的临床表现为心内膜纤维化引起的限制性心肌病,出现该病的患者由于心脏瓣膜病变,可致血液反流、心内血栓形成。部分患者可出现动、静脉血栓[13,14,15]。伴嗜酸性粒细胞增多及PDGFRα基因异常的髓系、淋巴系肿瘤患者实验室检查结果示:①患者血清类胰蛋白酶、维生素B12水平升高;②外周血中成熟嗜酸性粒细胞增多,该类细胞可出现细胞质中颗粒减少、空泡形成、细胞核过分叶或低分叶、体积偏大等异常改变,仅有少部分患者可见原始嗜酸性粒细胞增多[13];③中性粒细胞可增多,嗜碱性粒细胞及单核细胞多为正常[16],有时可出现贫血与血小板计数减少;④骨髓中嗜酸性粒细胞及其前体细胞增多,以成熟细胞为主,多不伴原始细胞增多。伴嗜酸性粒细胞增多及PDGFRα基因异常的髓系、淋巴系肿瘤患者骨髓活组织病理检查结果示:①呈纺锤体型的CD25+肥大细胞明显增多,且呈分散分布、松散簇状分布或簇状聚集样分布,骨髓网硬蛋白增多[17,18];②可出现骨髓坏死、夏科-雷登(Charcot-Leyden)结晶[15]。嗜酸性粒细胞常表达CD23,25及69[17];肥大细胞常表现为CD2–CD25+[18],有时表现为CD2–CD25–,偶尔表现为CD2+CD25+,而在系统性肥大细胞增多症(systemic mastocytosis,SM)患者中,肥大细胞多表现为CD25+,约2/3 SM患者同时表现为CD2+[9]。由于FIP1L1-PDGFRα为染色体4q12部分基因隐性缺失所致,携带FIP1L1-PDGFRα融合基因的患者细胞遗传学分析结果常显示为正常,所以需要通过分子生物学分析检测此融合基因存在与否[9]。
伴嗜酸性粒细胞增多及PDGFRβ基因异常的髓系肿瘤的临床表现:此类疾病多为MPN,如伴嗜酸性粒细胞增多的慢性粒-单核细胞白血病(chronic myelomonocytic leukemia,CMML),或伴嗜酸性粒细胞增多的不典型慢性髓性白血病(atypical chronic myelogenous leukemia,aCML),以及伴嗜酸性粒细胞增多的其他MPN[19,20];少数表现为AML(继发于原发性骨髓纤维化)与青少年型粒-单核细胞白血病(juvenile myelomonocytic leukemia)的血液学特征[21]。该类疾病患者主要临床表现为脾大,肝大者少见。部分患者存在皮肤浸润,由于心脏受损也可能会出现心衰。血清类胰蛋白酶水平存在轻至中度升高[9]。外周血中性粒细胞、嗜酸性粒细胞及单核细胞增多;嗜碱性粒细胞增多较为少见[22];可伴有贫血、血小板计数减少。骨髓有核细胞增生活跃,主要为中性和嗜酸性粒细胞增多;骨髓活组织病理学检查结果示呈纺锤体型肥大细胞增多[22,23],可见骨髓网硬蛋白增多[22]。在疾病慢性阶段,骨髓和外周血中原始细胞比例小于20%。增多的肥大细胞常表现为CD2+CD25+[22]。患者遗传学和分子学分析结果可见t(5;12) (q31-q33;p12)及ETV6-PDGFRβ融合基因[24],涉及5q31-33的其他基因重排也可形成其他包含PDGFRβ的融合基因。
伴嗜酸性粒细胞增多及FGFR1基因异常的髓系、淋巴系肿瘤的临床表现:该类疾病既往被称为8p11骨髓增殖综合征(8p11 myeloproliferative syndrome),由Macdonald等[25]于1995年正式提出,强调了位于8p11的FGFR1基因在此类疾病发生中的重要性。该类疾病的临床表现或为淋巴瘤的临床表现,如淋巴结肿大;或为骨髓增殖性疾病的临床表现,如脾大与高代谢综合征;也可为AML或者髓系肉瘤的临床表现,如发热、体质量减少及盗汗等[19]。该类疾病患者中,约90%存在外周血及骨髓中嗜酸性粒细胞增多[26]。嗜碱性粒细胞增多较为罕见,但在BCR-FGFR1融合基因呈阳性的患者体内,可出现嗜碱性粒细胞增多[27]。另有研究发现,在t(6;8)/FGFR1OP-FGFR1融合基因呈阳性者中,可出现真性红细胞增多症的临床表现[28]。
2008年,WHO关于CEL-NOS的诊断标准如下:①嗜酸性粒细胞计数≥1.5×109/L;②无Ph染色体,BRC-ABL融合基因,或其他MPN(真性红细胞增多症及原发性血小板增多症、原发性骨髓纤维化等),或MDS/MPN(CMML或aCML)的证据;③无t(5;12)(q31-35;p13)或其他涉及PDGFRβ的基因重排;④无融合基因FIP1L1-PDGFRα或其他涉及PDGFRα的基因重排;⑤无FGFR1基因重排;⑥外周血及骨髓中原始细胞比例少于20%,无inv(16)(p13q22)或t(16;16)(p13;q22)或AML的诊断特征;⑦存在细胞遗传学与分子生物学异常克隆的证据,或外周血中原始细胞比例>2%、骨髓中原始细胞比例>5%。该诊断标准一方面将嗜酸性粒细胞增多伴PDGFRα、PDGFRβ、FGFR1基因重排的病例排除在外,同时也排除了常见的伴嗜酸性粒细胞增多的髓系、淋巴系肿瘤;另一方面,又需要有嗜酸性粒细胞克隆性增多的证据。但是,嗜酸性粒细胞增多而不涉及PDGFRα、PDGFRβ、FGFR1融合基因及非酪氨酸激酶相关染色体异常的患者,在所有克隆性嗜酸性粒细胞增多的患者中所占比例<3%,同时,原始细胞增多也仅见于极少部分非选择性嗜酸性粒细胞增多的患者中[10]。因此,依据WHO诊断标准诊断的CEL-NOS病例,是非常罕见的[10]。由于既往难以鉴别诊断CEL-NOS与IHES,故CEL-NOS的实际发病率尚不清楚[7]。在CEL-NOS中,尚未发现明确的细胞遗传学或分子生物学异常,嗜酸性粒细胞增多伴PDGFRα、PDGFRβ、FGFR1基因重排的病例目前被排除在外。截至目前,在伴有嗜酸性粒细胞增多的MPN或MPN/MDS中,已发现50多种编码异常酪氨酸激酶的融合基因(图1)。通常认为,CEL-NOS可能是携带JAK2、ABL1或FLT3基因的一种亚型[10]。即使嗜酸性粒细胞增多与髓系肿瘤标志性染色体异常同时出现,尚不能认为增多的嗜酸性粒细胞源于克隆性增殖,因为在髓系肿瘤中也可出现继发性嗜酸性粒细胞增多[9]。
对于继发性嗜酸性粒细胞增多的患者,若积极治疗原发病,嗜酸性粒细胞可恢复至正常水平。对于淋巴细胞变异型HES患者,给予甲泼尼龙(15 mg/d)联合环孢素A(150 mg/d)治疗,可降低嗜酸粒细胞计数,改善临床症状[5]。对于特发性嗜酸性粒细胞增多患者,可采用泼尼松、羟基脲、α干扰素作为一线治疗方案。而对于上述治疗药物治疗效果差的患者,可尝试使用抗白细胞介素(interleukin,IL)-5的抗体美泊珠单抗(mepolizumab)或抗CD52单抗阿仑珠单抗(alemtuzumab)治疗,但是这2种药物在特发性嗜酸性粒细胞增多患者中的治疗效果,有待进一步研究。针对药物治疗后复发的特发性嗜酸性粒细胞增多患者,可考虑采用异基因造血干细胞移植(allogeneic-hematopoietic stem cell,allo-HSCT)[4]。
伊马替尼治疗FIP1L1-PDGFRα融合基因呈阳性的患者,具有较好疗效。相关研究结果显示,发病时间小于3个月的FIP1L1-PDGFRα 融合基因呈阳性的患者接受伊马替尼治疗,90%~95%患者可获得持久的完全血液学缓解(complete hematologic remission,CHR);分子学检测结果显示,发病时间小于12个月的患者接受伊马替尼治疗,80%~95% FIP1L1-PDGFRα融合基因呈阳性的患者可以获得完全分子学缓解(complete molecular remission,CMR),实现较高比例的无疾病进展生存和较高的总生存率[10]。截至目前,根据非正式共识,对于FIP1L1-PDGFRα融合基因呈阳性的患者,伊马替尼治疗的初始剂量为100 mg/d,达到CHR或CMR后,可将剂量调整为:①3次/周,100 mg/次;或隔天给药,100 mg/次;②1次/周,100~200 mg/次。这2种方案均可以实现持久CHR和CMR,且毒副作用低[10]。相关研究并未发现此类患者原发性耐药,而继发性耐药与FIP1L1-PDGFRα T674I[29]、FIP1L1-PDGFRβ D842V[30]突变有关。近期一项研究发现,帕纳替尼(ponatinib)对于引起继发性耐药的上述2种基因具有明显抑制作用[31],但实际临床应用价值还有待进一步研究证实。对于PDGFRβ基因突变患者,应用伊马替尼也可获得较好治疗效果[9]。也许由于携带PDGFRβ基因的患者与髓系肿瘤的关系更加密切,这类患者给予伊马替尼400 mg/d治疗,有望取得治愈[10]。目前,对于嗜酸性粒细胞增多伴FGFR1重排的MPN,尚无确切有效的酪氨酸激酶抑制剂,可以考虑应用干扰素、造血干细胞移植进行治疗[9]。
CEL-NOS的治疗:由于尚不能明确鉴别诊断CEL-NOS与IHES,在一些可能包含CEL-NOS的IHES相关研究中,所有患者5年生存率可达到80%[9]。羟基脲、α干扰素可用于初治或激素治疗失败的患者,其他细胞毒性化疗药物及造血干细胞移植均可用于治疗CEL-NOS[32]。
虽然随着分子生物学、免疫学技术在嗜酸性粒细胞增多相关疾病中的不断应用,临床对于此类疾病的诊治取得了不少进步。但是,目前此类疾病的分类、命名仍较为混乱,治疗效果也不甚理想。探索此类疾病预后相关的预测因子,以及如何更加快速、准确地区分继发性、克隆性和特发性嗜酸性粒细胞增多,进而选择行之有效的治疗手段仍存在较大困难。

























