
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Rabson-Mendenhall综合征是一种极为罕见的临床综合征,是由于胰岛素受体基因突变所导致,临床可表现为皮下脂肪减少、黑棘皮病、生殖器增大、多毛症、牙齿发育不良、面容特殊、重度高胰岛素血症等。其发病率极低,迄今为止文献报道仅10余例。为提高对本病的认识,我们对2011年9月于首都医科大学附属北京儿童医院内分泌科收治的1例Rabson-Mendenhall综合征病例进行了总结,并对相关文献进行了复习,以期提高临床医师对Rabson-Mendenhall综合征的认识。
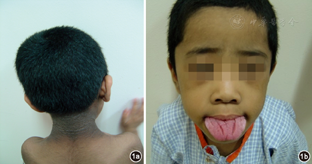
患儿男性,5岁5个月,因发现"皮肤黑伴多毛5年余"入院。患儿生后即表现全身皮肤色黑,以颈部、双侧腋窝、肘窝、腹股沟、腘窝为著,伴皮肤多毛,主要分布在颈背部、四肢。无发热、抽搐、呕吐、腹胀,无多饮、多尿、消瘦等症状,曾多次到当地医院就诊,一直未能确诊。患儿2011年9月于我院内分泌科就诊并收入院治疗。患儿为第二胎第一产,足月、因胎位不正行剖腹产,有宫内窘迫史,生后无窒息。出生体重3.1 kg。患儿生后4个月会抬头,6个月会坐,1岁时会走和会叫爸爸妈妈。生后4个月出牙,尚未换牙。母亲孕产史:第一胎:孕8周因胎停育行人工流产。第三胎,孕5周因劳累自然流产。第四胎第二产,男孩,2岁,体健,肤色正常。患儿父母非近亲结婚,均体健,肤色正常,无糖尿病家族史。入院后查体:身高106 cm,位于身高标准曲线第3百分位线,体重17 kg。营养欠佳,体形消瘦、匀称。全身皮肤可见黑棘皮伴多毛(图1a),主要分布于颈部、背部及四肢、双腋下、肘窝、腹股沟、腘窝、脐部。全身皮肤粗糙、干燥,皮下脂肪菲薄,皮肤松弛。头发浓密、卷曲,紧贴头皮生长;眼距宽,鼻梁低,鼻翼宽而厚。口大,唇厚外翻,口腔黏膜湿润,舌胖大,可见沟状分裂(图1b),牙列不齐。耳大、耳位低。心前区可闻Ⅱ级收缩期杂音。肝脾无肿大。四肢无明显畸形,手指及脚趾甲正常,无增厚表现。脊柱生理弯曲变浅。阴茎长6 cm,周径6 cm。睾丸容积5 ml,无阴毛、腋毛。入院后相关化验检查:口服葡萄糖耐量试验(OGTT)提示血糖正常,各时段胰岛素、C肽水平明显升高,超过检测的高限值(表1)。胰岛素自身抗体、胰岛细胞抗体、谷氨酸脱羧酶抗体均阴性。糖化血红蛋白:5.2%。胰岛素样生长因子-1<25.0 ng/ml(50~2986 ng/ml),IGFBP3为0.83 μg/ml (1.1~5.2 μg/ml)。性激素6项、皮质醇、甲功5项均正常。血常规、尿常规、肾小管系列、血生化全项、血氨、乳酸、尿筛查等均正常。生长激素激发试验正常。骨龄4岁,甲状腺、腹部及肾上腺B超均未见异常。肾上腺CT:肾脏增大,密度欠均匀。垂体磁共振成像(MRI):松果体区可见囊状长T1长T2信号,大小约14.5 mm×9.1 mm×9.3 mm,垂体高约4.2 mm,垂体内信号欠均匀,诊断:松果体囊肿,腺样体肥大。心脏彩超:先天性心脏病:室间隔缺损(嵴下),房间隔缺损(继发孔)。视力及眼底检查未见异常。智力测验:智力发育临界状态。皮肤活检病理回报:(腋下)送检皮肤,表皮角化过度,棘细胞层不规则增厚,表皮疣状突起,基底层无色素沉着,真皮乳头向上突起,突起乳头顶部及侧面表皮变薄,真皮浅层小血管周散在淋巴细胞浸润,符合黑棘皮病。诊疗经过:入院后根据患儿生后即出现症状,临床有皮肤黑棘皮样变,皮肤增厚且多毛,皮下脂肪减少,皮肤干燥伴多毛,生长发育迟缓,头发浓密、卷曲且紧贴头皮生长,眼距宽,鼻梁低、鼻翼宽而厚,口大、唇厚外翻,舌胖大、可见沟状分裂,牙齿排列不齐,耳大、耳位低,外生殖器增大等表现,临床诊断为Rabson-Mendenhall综合征。虽然患儿有明显的黑棘皮病表现,胰岛素水平明显升高,但是血糖正常,年龄仅5岁,故未给予二甲双胍等特殊治疗。黑棘皮病变部位外涂润肤油,皮肤干燥和角化改变好转。出院后随访,血糖及HbA1c水平正常。
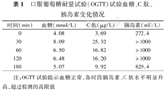
口服葡萄糖耐量试验(OGTT)试验血糖、C肽、胰岛素变化情况
口服葡萄糖耐量试验(OGTT)试验血糖、C肽、胰岛素变化情况
| 时间(min) | 血糖(mmol/L) | C肽(μg/L) | 胰岛素(mU/L) |
|---|---|---|---|
| 0 | 4.08 | 3.69 | 272.4 |
| 30 | 8.09 | 25.32 | >1000 |
| 60 | 6.50 | 16.82 | >1000 |
| 120 | 6.48 | 16.20 | >1000 |
| 180 | 5.07 | 9.92 | 829.4 |
注:OGTT试验提示血糖正常,各时段胰岛素、C肽水平明显升高,超过检测的高限值
胰岛素受体(INSR)基因[1]位于人常染色体19q13.2-13.3,几乎分布在人体内所有细胞的细胞膜上,是由2对α和β亚单位构成的四聚体。胰岛素与INSRα亚单位结合后,解除对β亚单位的负调控效应,β亚单位膜内结构域发生磷酸化,而发挥胰岛素的生物效应。Kahn等[2]1976年研究了6例严重胰岛素抵抗伴有黑棘皮病的妇女,根据患者发病特点、131I标记胰岛素和INSR结合情况,将严重胰岛素抵抗分为A型、B型。其中A型胰岛素抵抗是由于INSR基因缺陷所致,B型胰岛素抵抗是由于患者体内存在INSR抗体所致。在随后的研究中,人们陆续发现了其他严重的胰岛素抵抗综合征:如矮妖精貌综合征、Rabson-Mendenhall综合征等,上述综合征均属于A型胰岛素抵抗[1],它们共同的临床特征性表现为重度胰岛素抵抗,临床表现由重到轻。另外,还有部分INSR异常的病例,临床表现轻于上述综合征,且缺乏上述综合征的典型体征表现,而被一并诊断为A型胰岛素抵抗综合征。在一些伴有脂肪萎缩的2型糖尿病患者中,也发现有严重的胰岛素抵抗。故根据严重胰岛素抵抗的分子机制[3]又分为两大类,即:原发于INSR和受体后信号缺陷的严重胰岛素抵抗[致病基因如INSR和蛋白激酶B基因(Akt)],继发于全身或局部脂肪萎缩的严重胰岛素抵抗[致病基因如核纤层蛋白A基因(LMNA)、过氧化物酶体增殖剂活化受体-G(PPAR-G)等]。
本报告中,患儿为学龄前男童,生后即起病,病史长,以皮肤黑伴多毛为主要临床表现。入院后查体可见患儿全身皮肤色黑伴多毛,颈部、双腋下、肘窝、脐部、腹股沟及腘窝可见明显黑棘皮;左腋下皮肤活检病理提示符合黑棘皮病,故诊断黑棘皮病成立。患儿无明显多饮、多尿、多食、体重下降及抽搐,营养欠佳,体形消瘦。入院后行OGTT并C肽及胰岛素释放试验,患儿血糖水平尚处于正常水平,胰岛素和C肽水平则明显升高,显著高于正常值,提示患儿存在严重的高胰岛素血症。近年来对2型糖尿病和代谢综合征的研究,加深了我们对高胰岛素血症和黑棘皮之间相互关系的了解。同时,也将黑棘皮病和高胰岛素血症联系在一起。黑棘皮已经被认为是高胰岛素血症的体表标志。黑棘皮病常常发生在长期肥胖的人群。但是,肥胖导致的黑棘皮病常常发生在肥胖以后,且黑棘皮病有一个由轻症逐渐进展的过程。由于本患儿生后起病,我们推测患儿的病变很可能是先天性疾病。
通常黑棘皮病分为良性黑棘皮病和恶性黑棘皮病[4]。恶性黑棘皮病多于成年期发病,伴有不同脏器的肿瘤,多数为腺癌,以胃肠道、肺和乳腺多发。黑棘皮病可以出现在肿瘤发生前,也可以发生在肿瘤的晚期。良性黑棘皮病包括真性良性黑棘皮病、假性良性黑棘皮病和与黑棘皮相关的综合征。假性良性黑棘皮病多与肥胖症、库欣病等内分泌疾病相关。黑棘皮病的临床症状可以随内分泌疾病的改善而好转。与黑棘皮病相关的综合征包括Rabson-Mendenhall综合征、矮妖精貌综合征、Lawrence综合征、Rud综合征和Berardinelli-Seip综合征等。
Rabson-Mendenhall综合征1956年[5]由Rabson和Mendenhall首次报告并命名。本综合征是一种由INSR基因突变所导致的、具有多种特征性躯体异常的遗传性疾病,表现为严重胰岛素抵抗和黑棘皮病。其遗传方式为常染色体隐性遗传,多为INSR复合杂合突变所致。Rabson和Mendenhall最初的报告来源于3位同胞兄妹,包括一位男性和两位女性。遗传方式均为常染色体隐性遗传。Rabson-Mendenhall的主要表现有严重胰岛素抵抗综合征的表现[6]:包括明显高胰岛素血症和胰岛素抵抗、黑棘皮病、高雄激素血症,根据患者病程进程的不同可以表现为低血糖,也可以进展为糖尿病;特征性的皮肤改变,包括黑棘皮、多毛症、皮下脂肪发育不良;特殊的面部特征包括:丘比特弓形唇,唇厚,低鼻梁,舌胖大且舌面有裂隙,牙列不齐,大耳朵;此外具有一些特殊的伴随症状:出牙早,生长发育迟缓、厚指甲,外生殖器增大或青春发育提前,松果体增生和肾脏异常。成人女性患者可由于高胰岛素血症继发的高雄激素血症,导致多囊卵巢、闭经、男性化等表现。因属于先天性疾病,常于儿童发病,大部分可以存活到青春期[7],存活的时间决定于INSR突变后残存的INSR结合能力。随着医疗水平的提高,患者的存活时间也得以延长,近期文献报告一例患者已经存活达到45岁,但是已经因终末期肾功能衰竭行肾移植[8]。
本报告中患儿具有典型的Rabson-Mendenhall综合征皮肤改变,严重胰岛素抵抗的表现,本综合征特殊的面容表现,以及本综合征常见的伴随症状,均与文献报告的临床表现一致,故临床诊断为Rabson-Mendenhall综合征。与文献报告不同的是本患儿没有明显的指甲增厚。我们还观察到患儿的皮肤松弛易牵拉,关节韧带松弛,这些表现是文献中没有报告的。
本病诊断主要应与矮妖精貌综合征、脂肪萎缩综合征、B型胰岛素抵抗及恶性黑棘皮病进行鉴别。矮妖精貌综合征又称Donohue综合征,为INSR最严重的一种病变,见于INSR基因的纯合和杂合突变,典型特征为宫内发育迟缓、特征性的鸟样("小妖精"样)面容、严重胰岛素抵抗、空腹低血糖症,多于生后2年内死亡。两者临床表现很相似,如都有特殊容貌、生长发育异常、皮下脂肪减少等,但Rabson-Mendenhall综合征有其特征性表现:出牙早且伴牙列不齐、指(趾)甲增厚、松果体肥大等,临床表现不如Donohue综合征严重,生存期相对长。两者临床表型严重程度不同最根本的原因在于INSR基因突变残存的受体功能的不同。Donohue综合征INSR的基因突变通常位于细胞膜外α亚基,直接影响胰岛素与受体的结合,INSR的结合能力通常小于正常对照的10%[9],甚至是INSR受体后酪氨酸磷酸化活性的完全丧失[10];而Rabson-Mendenhall综合征INSR基因突变多位于胞内β亚基,受体结合能力可以达到正常对照的10%~30%[11]。脂肪萎缩综合征,包含了一组遗传学异质性的罕见综合征,临床表现为对胰岛素不敏感和皮下缺乏脂肪组织,可以发生胰岛素抵抗性糖尿病,伴有局部或全身性皮下脂肪组织减少或缺失和高脂血症。根据遗传方式分为先天性和获得性,根据范围不同可有全身性和不同类型局部性脂肪萎缩。先天遗传型的常与PPAR-γ、LMNA等基因突变有关。通常获得性脂肪萎缩是由于后天因素所致,非INSR基因突变所致。B型胰岛素抵抗综合征[12]是血循环中存在针对胰岛素受体抗体的一种自身免疫性疾病。40~60岁女性多见,女∶男为2∶1,多伴有其他自身免疫性疾病,1/3患者有系统性红斑狼疮或干燥综合征。症状时轻重不等,临床的严重程度随循环自身抗体滴度和性质的变化。此类疾病患者的INSR正常。1990年,Kadowaki等[13]首次发现并报道了与Rabson-Mendenhall综合征相关的INSR突变,之后陆续报道的INSR缺陷包括错义突变、无义突变、插入、缺失突变及复合重排等[14]。2007年,Tuthill等[11]报告了2例同胞姐妹同患Rabson-Mendhall综合征,基因突变分析显示:该2例患儿均携带两个INSR的复合杂合突变,分别为Arg209His的功能丧失突变及配体结合区的Gly359Ser突变。这两个位点的突变分别来源于父亲和母亲,均为杂合突变。Arg209His纯合突变也可见于矮妖精貌综合征。2009年,国内蒋怡然等[15]曾报道一例Rabson-Mendenhall综合征,患者INSR第6号外显子区发生了Met469Thr突变,突变基因来源于父亲,患儿母亲该位点未发现异常。Jiang等[16]报告了一例11岁女性患儿为复合杂合突变,其携带的Arg83Gln (R83Q)突变使INSR表达减少,其胰岛素结合后酪氨酸磷酸化减少,IRS-1、Akt和Erk1/2分别降低到野生型的60%、40%和50%,提示该突变影响了INSR受体后信号转导。本患儿的基因突变类型尚不明确,有待进一步的遗传学研究来证实。
INSR基因的突变可能影响INSR结构和功能的许多环节,目前已经发现60余种INSR基因突变。Taylor等[17]曾将INSR所致的功能改变进一步分类,主要包括:INSR合成mRNA减少;受体翻译后转运至细胞膜表面发生障碍;受体对胰岛素结合的亲和力下降;受体β亚单位自身磷酸化和酪氨酸激酶活降低;INSR降解加速等。由于INSR突变,导致胰岛素不能与INSR正常结合,或结合后不能发挥正常的生理效应,导致发生胰岛素抵抗。机体通过胰岛素细胞的过度分泌、或者胰岛细胞的增生,增加胰岛素的分泌量进行代偿,使血糖水平保持正常。长期胰岛素的超负荷分泌,将导致胰岛细胞功能衰竭,或过量分泌的胰岛素仍不足以代偿以降低血糖,从而导致糖尿病的发生。过高的胰岛素通过残存的INSR和(或)胰岛素样生长因子受体,促进皮肤角朊细胞和真皮母细胞的增殖,导致黑棘皮病和指甲增厚。INSR基因突变还可以抑制与代谢、生长相关基因的表达,这可能是其导致生长发育落后的机制之一。
总之,Rabson-Mendenhall综合征有着复杂的临床特征及遗传发病机制。由于临床极为少见,临床极易误诊。临床遇有严重胰岛素抵抗并伴发有相关临床表现的患儿应考虑到本综合征的可能性。

























