
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Galloway-Mowat综合征作为一种累及多系统的临床综合征,以肾脏、神经系统受累居多,神经系统畸形影像学表现多样。Dandy-Walker综合征是一种先天性脑发育异常性疾病,以神经系统发育畸形为主要表现。目前国内尚无神经系统畸形表现为Dandy-Walker综合征的Galloway-Mowat综合征的病例报道,现综合相关文献,对首都医科大学附属北京儿童医院收治的1例患儿进行分析,以加强对该类疾病的认识。
患儿,女,3岁,因"眼睑水肿20 d,稀便1 d"入院。患儿入院前20 d无明显诱因出现晨起双眼睑水肿,家长未予重视,无特殊治疗。入院前13 d,患儿双眼睑水肿加剧,伴腹部膨隆,就诊于当地医院,查尿常规+++,总胆固醇17.75 mmol/L,考虑肾病综合征,予口服泼尼松2 mg/kg,实际给予27.5 mg/d;入院前1 d出现发热,体温最高38.1 ℃,伴黄色稀水样便,水肿无明显改善,并出现尿量减少,小便2次/d,遂就诊于首都医科大学附属北京儿童医院肾内科。发育状况:患儿4个月抬头,8个月会坐,1岁6个月可扶走,3岁不会说话,仍需扶走。查体:血压100/50 mmHg(1 mmHg=0.133 kPa),头围46 cm,特殊面容(发际低,鼻梁宽扁,眼裂小,上唇长,颈短),行走不稳,眼睑及面部轻度水肿,咽充血,扁桃体Ⅰ度肿大,双肺呼吸音粗,未闻及啰音,心脏查体未及异常,腹膨隆,腹壁轻度水肿,腹围53 cm,移动性浊音可疑阳性,余腹部查体未及异常,脑膜刺激征及病理征阴性,双下肢轻度水肿。尿常规:蛋白+++,潜血++,镜检红细胞2~4个/高倍视野(HP),镜检管型1~2个/HP;血常规:白细胞21.4×109/L,分类中性粒细胞78%,血红蛋白150 g/L,血小板441×109/L;大便常规:黄色稀便,白细胞2个/HP,轮状病毒阳性。入院生化检查:总蛋白34.0 g/L,白蛋白11.9 g/L,胆固醇15.87 mmol/L,血肌酐、尿素氮正常,24 h尿蛋白定量155.75 mg/kg。入院前行头颅MRI:弥散性巨脑回畸形并白质发育不良(图1),小脑发育不良并后颅窝脑脊液增多(图2),符合Dandy-Walker变异型。染色体核型分析为46,XX;无其他异常。Gesell儿童智力发育诊断表:适应能力和精细动作能力轻度低下,大运动能力、语言能力和个人社交能力中度低下。结合病史及辅助检查结果诊断为肾病综合征(肾炎型);轮状病毒性肠炎;精神发育迟缓;Dandy-Walker综合征。予抗感染、对症止泻等治疗后,腹泻好转。继续足量泼尼松(2 mg/kg)口服,病情好转后带药出院。足量激素口服8周,期间间断给予甲泼尼龙冲击治疗3次,足量激素口服第4周起加用环孢素(3 mg/kg)口服,2周后调整剂量为5 mg/kg,多次复查尿常规提示尿蛋白+++,再次收住院行肾脏穿刺检查,结果:可见34个肾小球,其中1个球性硬化,余肾小球系膜细胞和基质轻度弥散增生,局灶、节段性中重度增生,其中14个节段性硬化伴灶状足细胞增生、肥大;肾小管上皮空泡及颗粒变性,灶状萎缩,肾间质灶状淋巴、单核细胞浸润,伴纤维化,小动脉无明显病变。免疫荧光:IgM(+),补体1q(±),IgA、IgG、补体C3、补体C4、Fib均阴性。电镜:肾小球系膜细胞和基质轻度增生,系膜区可见块状电子致密物沉积,基底膜未见病变,上皮足突可见融合,符合局灶节段性肾小球硬化症(NOS型)。
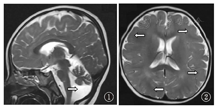
MRI shows pachygyria malformation
MRI shows cerebellar hypoplasia and cystic dilatation of the fourth ventricle
MRI shows pachygyria malformation
MRI shows cerebellar hypoplasia and cystic dilatation of the fourth ventricle
Galloway-Mowat综合征是一种常染色体隐性遗传病,临床表现以肾病综合征伴精神运动发育落后为主。该病于1968年由Galloway与Mowat等首次报道,多于3岁以内起病,可伴小头畸形、特殊面容、视力和听力障碍以及骨骼发育异常[1]。神经系统受累临床表现复杂,影像学特点多样。既往报道的Galloway-Mowat综合征患者神经系统畸形可表现为小脑发育不全、小脑萎缩、脑发育不良、皮质萎缩、脑白质软化、颞叶及海马萎缩、脑穿通畸形、髓鞘发育不良或减少、巨脑回、多小脑回及第四脑室扩张等;亦有报道伴神经系统受累临床表现但无神经系统影像学改变的病例[2,3,4],临床可表现为癫 、精神发育迟缓、下肢/四肢轻瘫及运动发育落后于同龄儿。肾脏病理以NSO型表现居多,也可表现为弥散性系膜硬化、微小病变及系膜溶解[2,3,4],因此多数病例对于激素耐药并最终发展为肾衰竭。本例患儿为肾病综合征(肾炎型),伴精神发育迟缓、特殊面容及小头畸形,MRI提示存在巨脑回畸形,小脑发育不良及脑积水,符合Galloway-Mowat综合征诊断。
、精神发育迟缓、下肢/四肢轻瘫及运动发育落后于同龄儿。肾脏病理以NSO型表现居多,也可表现为弥散性系膜硬化、微小病变及系膜溶解[2,3,4],因此多数病例对于激素耐药并最终发展为肾衰竭。本例患儿为肾病综合征(肾炎型),伴精神发育迟缓、特殊面容及小头畸形,MRI提示存在巨脑回畸形,小脑发育不良及脑积水,符合Galloway-Mowat综合征诊断。
Dandy-Walke综合征是一种以第四脑室和小脑发育障碍为特征的少见的中枢神经系统发育畸形,通常不伴有其他系统受累表现,目前其病因尚不明确,可能与感染、遗传或代谢等因素有关。1914年由Dandy和Blackfan首先报道,其发生率为1/25 000~1/30 000妊娠,占婴幼儿脑积水的3.5%。临床可分为典型和变异型2类,后者相对前者临床表现轻,预后相对较好。典型Dandy-Walker畸形的三联征包括小脑蚓部部分或全部发育不全、第四脑室囊状扩张及后颅窝扩张引起横窦、小脑幕和窦汇上移[5]。本例患儿神经系统畸形表现为弥散性巨脑回畸形并白质发育不良,小脑发育不良并后颅窝脑脊液增多,符合Dandy-Walker综合征(变异型)诊断,该特点在既往Galloway-Mowat综合征中尚无报道。部分病例报告中,Galloway-Mowat综合征患儿神经系统损害出现早于肾脏损害,本例患儿也是肾脏损害出现较晚,提示对于Dandy-Walker综合征患儿,应注意监测尿常规,观察有无合并肾脏受累情况,可减少临床漏诊。
Galloway-Mowat综合征与Dandy-Walke综合征神经系统畸形表现存在交叉时,临床易混淆或漏诊,尤其是部分Galloway-Mowat综合征患儿肾脏受累表现轻或者出现较晚时。由于致病基因定位尚不清楚,基因学诊断在Galloway-Mowat综合征的应用尚缺乏经验,但已知3号、9号及13号染色体异常与Dandy-Walke综合征发病有关,并在多个家系中检测到不同的突变基因,如层黏连蛋白r-1基因和巢蛋白-1基因等[6]。因此有待进一步基因学研究来辅助诊断。本例患儿虽染色体核型分析未见异常,家族中也无相似疾病患儿,但Galloway-Mowat综合征属于常染色体隐性遗传,同胞患病的危险性为1/4,本例患儿无同胞兄弟姐妹,因此建议对患儿父母进行遗传咨询,如再次妊娠孕期超声检查应注意胎儿神经系统及肾脏发育情况,并监测胎儿有无生长缓慢、脑积水、双顶径偏小等情况,对宫内诊断该病有一定帮助。
本例肾脏病理表现为局灶节段性肾小球硬化症(FSGS),该病理类型因其不良预后为人们所关注,FSGS患儿激素治疗有效者仅约占25%,余为部分有效或耐药,患儿常逐渐进入慢性肾衰竭。有报道对75例患儿随访7~217个月,8例(11%)完全缓解,17例(23%)肾小球滤过率(GFR)减退,16例(21%)已进入终末期肾(ESRD)[7]。本例患儿规律应用足量泼尼松2 mg/kg,口服8周后尿蛋白持续+++,为激素耐药型肾病综合征,提示远期预后不良。环孢素是FSGS二线治疗药物,环孢素联合甲泼尼松冲击(PMT)治疗FSGS患儿有一定疗效。Walao等[8]采用环孢素联合甲泼尼松冲击隔日共2周,再每周1次应用6周,第3周起加口服泼尼松2 mg/kg(隔日)及环孢素6 mg/kg治疗10例,其中8例完全缓解,1例部分缓解,仅1例进入ESRD。但有报道应用环孢素联合泼尼松治疗缓解者在52周时有40%复发,78周时60%复发[9]。本例患儿应用PMT联合环孢素A治疗,多次复查尿常规示尿蛋白+++,治疗无缓解。
本例患儿临床表现为肾病综合征,同时伴有小头畸形、特殊面容以及脑发育异常,肾脏为多系统受累表现之一。临床上肾病综合征合并有其他系统畸形时应注意与下列疾病相鉴别。(1)Pierson综合征[10]:临床表现为先天性肾病综合征、视觉系统发育异常(瞳孔、角膜、视网膜、视力),神经系统受累可表现为肌无力及智力异常,由于LAMβ2基因突变导致肾脏板层素β2缺乏致病,多于5岁内死亡。(2)Schimke免疫-骨发育不良[11]:以肾病综合征、脊柱骨骺发育不良、T淋巴细胞免疫缺陷及特殊面容为主要临床表现,致病基因为SMARCAL1基因,多15岁前死亡,个别可存活至成年。(3)Denys-Drash综合征[12]:临床表现为肾病综合征伴男性假两性记性、Wilms瘤或二者之一,由于Wilms瘤抑制基因(WT1基因)突变致WT1蛋白结构改变,造成DNA转录过程异常致病,3岁以内可进展为ESRD。(4)Frasier综合征[13]:以肾病综合征、男性假两性畸形以及性腺肿瘤为特点的临床综合征,由WT1基因突变导致WT1不同亚型蛋白质产物比例失衡致病,多于2~6岁起病,进展缓慢,可青春期后出现肾衰竭。(5)甲-髌综合征[14]:肾脏病变可表现为血尿、非肾病/肾病水平的蛋白尿以及肾衰竭,另外可伴有指甲发育不良,骨骼及视觉系统(虹膜、睫状体异常,青光眼、白内障等)异常,由于LXM同源盒转录因子1基因突变致功能缺失,影响中胚层及外胚层发育而致病,其预后取决于肾脏损伤情况。(6)Zellweger综合征[15]:临床表现为智力及运动发育迟缓或不发育、肝脏及肾脏功能异常,也可伴有骨骼、泌尿生殖腺以及视觉系统发育异常,由于过氧化物酶体基因突变致过氧化物酶体的代谢功能障碍,该病进展迅速,多1岁内死亡。

























