
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
多发性或单发骨纤维结构不良、皮肤色素沉着及外周性性早熟,是McCune-Albright Syndrome(简称MAS)临床表现的3个特点,也是临床确诊的全部依据[1]。此病在1936年及1937年被美国医生McCune[2]和Albright等[3]分别报告后被命名为McCune-Albright综合征。该征同时伴有不同程度、不同种类、变化多端的内分泌功能障碍,包括:性腺、甲状腺、脑垂体、肾上腺、甲状旁腺等,甚至还包括非内分泌系统的功能障碍如肾脏、肝脏病损,病情十分复杂[4]。国外流行病学调查其患病率为1/10万~1/100万之间,各种族均可发病,白种人相对多见,黄种人少见[5]。男女均可受累,女性患病率明显高于男性。多在童年期发病,也有新生儿期就发病的报导。该病临床少见,国内最早报告于1979年,国内外多以个案形式报告,目前尚未被多数临床医师认识[6,7,8,9]。对于MAS,目前尚无有效的根治疗法,主要采取对症治疗。其预后个体间差异较大,取决于受累器官的种类及程度。本文根据文献报道,就McCune-Albright综合征的病因、发病机理、临床表现、诊断及治疗等问题进行总结。
MAS的病因及发病机制现已基本阐明[4,5]。该病的关键遗传改变是在胚胎早期,单个细胞GNAS基因编码鸟嘌呤核苷酸结合蛋白(G蛋白)α亚基区域发生突变,导致G蛋白结构与功能异常。如果这一突变,发生在胚胎内细胞团期(胚胎干细胞期),则源自胚胎3个胚层的组织均可受累。伴随着胚胎发育,突变细胞形成的克隆散在分布于整个机体,形成突变嵌合体(即同一组织器官,可能同时存在突变细胞及正常细胞克隆),基于突变细胞累及的器官与组织,出现MAS的不同临床表现。
MAS发生突变的GANS基因位于第20号染色体长臂,编码G蛋白。G蛋白(包括α、β、γ亚基)是偶联细胞外激素(第一信使)与腺苷酸环化酶激活释放cAMP(第二信使)的关键蛋白。脑垂体多种激素,包括黑色素细胞刺激激素,促黄体激素,甲状腺刺激激素,生长激素刺激激素和肾上腺皮质刺激激素,其效应均通过G蛋白α亚基偶联腺苷酸环化酶体系而发挥作用。在MAS患儿,G蛋白的α亚基功能异常,靶细胞内腺苷酸环化酶组成性激活,不受机体负反馈机制调控,产生大量细胞内cAMP,致黑色素、雌二醇、睾酮、甲状腺素、生长激素和皮质醇等激素持续分泌增加。根据受累器官组织的不同,可出现牛奶咖啡斑、性早熟、骨纤维结构不良、肢端肥大症、甲状腺机能亢进以及库兴氏综合征等不同临床表现。此外,由于G蛋白的α亚基异常主要影响垂体激素在靶器官的效应,而在非内分泌器官,可能检出突变细胞但无相应临床表现。
是指女童在8岁前、男童在9岁前呈现第二性征的发育异常性疾病。外周性性早熟又称假性性早熟,由于卵巢自主分泌雌激素所致。外周性性早熟是MAS最早出现、最常见的内分泌异常,且常在婴幼儿期出现,在女童表现为不规律的周期性阴道流血。其次是第二性征提前发育,如乳腺发育、阴唇肥大、出现阴毛等。外周性性早熟,男性很少见,一旦发病,可表现为巨睾症、睾丸微石症,成年后可出现巨人症、肢端肥大症等[10,11,12]。
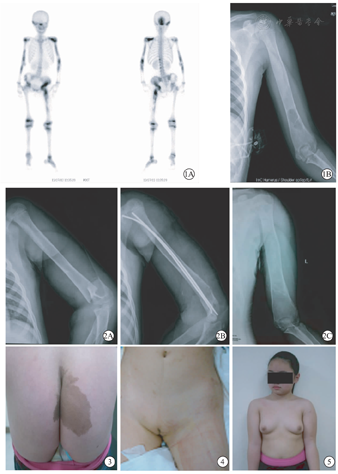
常表现为单发或多发性骨纤维结构不良,(fibrous dysplasia of bone,FD),是以骨的纤维组织增生,伴骨化不全为病理特点的良性骨肿瘤。多在儿童期发病,伴内分泌功能障碍时以多骨型更多见。骨受损出现时间较早,任何骨均可受累,有文献报道50%的骨损害出现在8岁左右[13]。多呈不对称性分布,颅骨、上下颌骨及股骨上干骺部,发生频率最高,其次脊柱、骨盆、长骨骨干、干骺端甚至骨骺等也有受累的报告(图1,图2,图3,图4,图5为作者亲自治疗的典型McCune-Albright综合征病例)[14]。FD发生在颅面骨时常病情发展缓慢,症状、体征不明显,但病变累及颅底并过度增生时,可导致脑神经孔受压,如视神经、听神经受压可致视听力下降甚至失明、失聪的严重后果。如上下颌骨受累可致鼻腔通气、上消化道吞咽功能受阻及颜面毁损,甚至受到生命威胁[15]。FD如发生在肢体骨骼,早期或轻型病变往往缺乏症状和体征,发生病理骨折后,才出现症状和体征,如疼痛、跛行、活动受限等[16]。骨折年龄在4~9岁最常见,随年龄增长,病变可趋向静止,但不包括头面骨。发生在脊柱的FD可以因脊柱后突侧弯,或椎体病理性压缩骨折而就诊,也有因病损向椎管内扩展发生脊髓、神经根受压症状如运动及感觉障碍、肢体瘫痪、患区疼痛等[17]。
除性腺外还涉及甲状腺、脑垂体、肾上腺、甲状旁腺等内分泌改变及肝肾改变。MAS病例有甲状腺功能亢进表现的占33%~38%,此外尽管并无甲状腺功能亢进表现,但甲状腺素血清学检查发现T3、T4均有增高,应予以定期监测[19]。如发现甲状腺出现孤立性病变,应进一步追查原因[20]。20%~25%的病例出现生长激素过度分泌,往往同时出现催乳素过量,可使头面部骨病损加重,导致视听功能障碍,应追查脑垂体形态与内分泌功能变化[21]。9%的病人出现肾上腺皮质功能亢进,可在新生儿期出现库欣综合征,有的可自然缓解,也有的因病情严重而死亡,应予以关注[22]。对这种病症的筛查从体检开始,其体征可显示满月脸、多毛症等,并可与实验室评估相结合,实验室检查包括尿液及唾液的皮质醇测定等,同时还应追查肾上腺有无形态变化[23]。原发性甲状旁腺功能亢进症罕见可能不是MAS的一部分,由于维生素D缺乏导致继发性甲状旁腺功能亢进症在FD和MAS病人中较常见,可导致FD病损加重。一旦发现应治疗。血清总钙和游离钙、甲状旁腺激素的测定是甲状旁腺功能亢进的必要评估手段。肾脏的磷酸盐消耗可致不同程度低磷酸盐血症,可出现佝偻病体征。肝脏胆汁淤积可见于伴有库欣综合征的新生儿中[22,24]。
到目前为止,临床三大表现仍然是MAS诊断的主要依据[4,5]。对不典型病例(指3种临床表现只有2种甚至1种),检测20号染色体长臂区,编码G蛋白α亚基基因突变可有助诊断[25,26]。MAS的基因诊断,尽管近年来从理论认识、技术手段、大宗病例分析等方面均取得长足进展,但由于该病基因突变发生于胚胎期,突变体细胞克隆伴随机体生长、细胞分化,散在分布于不同组织器官,形成突变嵌合体,单一组织检测,易得阴性结果,而阴性结果亦不能排除突变受累组织的存在。因此目前尚不能作为常规诊断方法。当前,外周血白细胞DNA检测技术可操作性强,国内外已较普及,但阳性率较低。除皮肤外的受累组织如卵巢、睾丸、骨骼、甲状腺等基因检测阳性率明显高于外周血白细胞;多组织联合检测可进一步提高阳性率,但组织获取、保存相对困难,限制了其临床应用。展望未来,随着科学技术的进步,探索更具靶向、精确及敏感性的基因检测技术,有望成为最有价值的确诊方法。
性早熟分两类即外周性和中枢性性早熟。外周性性早熟主要由性腺、肾上腺等内分泌器官及摄入外源性性激素所致,MAS是外周性性早熟重要病因之一。MAS女患儿其卵巢细胞因基因突变,在无促性腺激素刺激情况下促黄体生成素受体自动激活而发生自律性雌激素分泌过多并形成有功能的卵泡膜细胞促使性早熟。这种性早熟,不受下丘脑-垂体-性腺轴调控,这就是与中枢性性早熟的区别。外周性及中枢性性早熟临床上两者均表现为第二性征提早发育时可能难以区别。如较长期观察,可发现外周性性早熟,性征发育可为非进行性,生殖系性征发育相对较慢,甚至停止继续发育,而中枢性性早熟多为进行性,直至生殖系统发育成熟,不会停止发育。实验室检查:垂体兴奋实验阴性,可排除中枢性性早熟。有助于诊断外周性性早熟。要注意的是外周性性早熟,少数病例在一定条件下可转化为中枢性性早熟,除了通过临床表现帮助鉴别外,更有必要作垂体兴奋实验予以鉴别[27,28]。
骨骼FD病变的确诊除了上述临床表现外,骨的影像学检查对诊断和鉴别诊断意义重大。骨的影像学检查X线片上呈典型"毛玻璃样变",最常见于颅面骨及股骨近端,多见于儿童和年轻人,随着病程进展,年龄增大继而形成囊性变,在颅面骨出现骨硬化改变。而股骨上段表现为弯曲畸形外观呈典型牧羊人手杖,被公认为FD最具特色X线表现。其原因是病灶区骨化不全,骨质软,负重后容易变形所致。此外,ECT(静脉注射99mTc的全身核素骨显影)检查,虽然不具有特异性,但对发现FD的多发病灶,尤其对无症状的潜在病灶,有不可取代的诊断价值[29]。
长骨病变来自髓腔内并向外膨胀代替正常骨组织,导致骨皮质变薄并常伴有溶骨性透亮区(囊性变)与骨的嗜酸性肉芽肿、骨囊肿等有时不易区别,但经病理检查可以鉴别。当出现儿童脊柱后突、椎体病理性压缩骨折,或椎体、附件骨质破坏时,在国内除考虑脊柱结核外,FD的可能性不能排除,尤其如有牛奶咖啡斑体征,更应想到MAS的可能性。最后确诊也有赖于病理检查。
到目前为止,MAS并无根治方法,主要是对症处理。如何治疗,取决于受累组织类型和程度[1,5,32]。
抑制性发育应视为治疗原则。有作者提出用抑制性发育的药物如环丙孕酮,70~100 mg/d,也有用雌激素受体拮抗剂三苯氧胺(他莫昔芬),口服20 mg/d,对抑制性发育功能均有较好效果,但具体用药种类、剂量、时间、看法并不一致,尤其可能发生的副作用尚有待进一步探讨[33]。婴幼儿期发现卵巢囊肿,有自愈可能,不宜轻易切除。
对于颅面骨FD,绝大多数不需要手术干预,但可能发生进行性视、听力、颜面损害,要求经验丰富的神经外科、颌面外科、整形外科联合完成高难度的颅底手术。发生在肢体的FD,如无骨折、疼痛、功能障碍,可持续观察,暂不处理。有作者提出,当发生难以处理的疼痛时,二磷酸盐是有效止痛方法。用于抑制骨吸收的药物二磷酸盐,可改善骨代谢、增加骨形成,是否有效,有待进一步从理论和实践进行探讨[34]。此外对于四肢的FD,尤其是下肢骨,加强所在骨肌肉功能训练,尤其不负重的功能训练,如游泳、骑车等,可减少骨折风险,也是不容忽视的治疗手段。
原发性骨病变破坏了正常的骨结构,由于负重因素,儿童长骨较扁骨更易发生病理性骨折,如发生病理性骨折或有碍关节功能时应按外科原则进行治疗。对于肿瘤病灶,一般情况下采取彻底刮除处理,如果肿瘤病灶破坏严重,无法保持完整的骨干连续性,则考虑行瘤段切除、植骨重建手术。临床上骨病灶清除术后形成的骨缺损处理棘手,为保持骨结构的完整性和骨及邻近关节功能的稳定性,常用修复方法主要包括自体骨、异体骨、组织工程骨、人工骨植骨等。上述方式均非完美,自体髂骨植骨来源有限、取骨手术增加创伤,存在并发症风险;同种异体骨有免疫排斥、交叉感染、愈合缓慢等缺点;组织工程骨尚无儿童应用的报道;而含磷酸盐的人工骨虽有良好的生物相容性,但愈合和吸收相对慢。
对于病理性骨折,多数学者主张采取积极措施,以个体化的手术原则,合理利用内固定材料,同期行骨折复位,以恢复肢体的解剖结构及强度。内固定方式多样,根据具体病情可考虑选择弹性髓内针、钢板螺钉、克氏针等。选择钢板螺钉时与普通骨折应有不同,应选择足够长度的钢板,在骨折近、远端正常骨组织上进行坚强内固定后,再视情况用螺钉固定病灶区已植骨的长骨骨干;选择弹性髓内针时,先清除病灶,然后用髓内针恢复长骨轴线和对位,骨折断端基本稳定后再行病灶区植骨。对于儿童长骨病理性骨折,只要条件允许,弹性髓内针内固定操作简便,损伤小,不累及骨骺,复位良好,恢复快,便于早期康复,并发症少,二期取出时创伤小,值得临床推荐[35]。
牛奶咖啡斑无不适者,不必治疗。
应针对异常表现做出相应对症处理。
MAS预后差异较大,主要取决于病变骨受累部位和程度,及内分泌障碍程度和范围。受累于头面部的骨病损,如上下颌骨病变可导致上呼吸道阻塞,危及生命。脊柱病损可致脊神经脊髓受压症状。外周性性早熟其预后比中枢性性早熟好,性征变化并非必然进行性加重。长期随访发现,部分患儿成人后可获生育能力。只要保持长期监护和恰当处理,大多数预后良好,但少数转化为中枢性性早熟或其他严重内分功能障碍,或颅底骨FD增生、硬化导致颅脑神经受压者,预后不良。
总之,McCune-Albright综合征,以一系列临床症候群的表现而命名,并非单一病种,十分罕见,病情十分复杂,涉及内分泌科、妇产科、小儿内外科、骨科、神经外科、耳鼻咽喉外科等多专业,只有共同定期、动态随访,才能发现各种问题并得到正确及时的处理。

























