
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
周围神经病、共济失调和视网膜色素变性(neuropathy, ataxia and retinitis pigmentosa,NARP)综合征是由线粒体编码的ATP6蛋白基因突变导致的母系遗传的线粒体脑肌病,临床以周围神经病、共济失调、视网膜色素变性为主要表现。我们现报道1例NARP患者,对其临床特点、影像学表现、电生理特点、骨骼肌病理及ATP6基因进行分析,供临床参考。
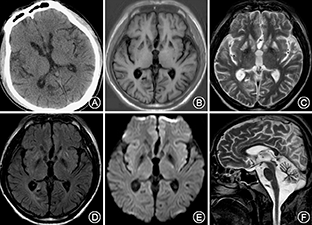
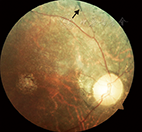
患者男性,38岁,因"头晕16年,走路不稳伴视物模糊12年,发作性意识丧失伴肢体抽搐8年,反应迟钝、全身无力3年"于2014年9月收入我科。2007年,患者曾因"脑外伤"昏迷数日,于外院进行治疗,曾被诊断为"脊髓小脑变性、齿状核-路易体-苍白球-红核变性"等疾病,行相关基因检测均未发现致病突变。否认其他既往病史,26岁结婚,育有1子,身体健康。父亲体健,母亲身材矮小,视力及听力均下降;2位姐姐及1位哥哥均体健。入院体检:体重51 kg,身高160 cm,一般状况可,体检欠配合。弓形足,内科系统体检无异常;意识清楚,反应迟钝,记忆力及计算力减退;双侧视敏度下降,左侧0.5,右侧0.3(国际标准视力表),视盘边界清,色暗黄,视网膜可见骨细胞样色素沉着,双耳听力下降,构音障碍;四肢肌张力略低,肌力Ⅳ级,腱反射减低;四肢振动觉略有减退,痛觉及关节位置觉尚可;自主运动缓慢,双侧指鼻试验欠稳准,双侧跟膝胫试验欠稳准,闭目难立征阳性,共济失调步态;双侧巴宾斯基征、双侧夏多克征阴性;脑膜刺激征阴性。一般实验室检查:同型半胱氨酸16.3 μmol/L,抗甲状腺球蛋白抗体205 U/ml,抗甲状腺过氧化物酶抗体59.67 U/ml,甲状腺球蛋白0.05 ng/ml,甘油三酯1.9 mmol/L,铜蓝蛋白672 U/L,余血液检查未见异常。脑脊液检查:脑脊液蛋白0.50 g/L,余未见异常。血乳酸检测:患者行乳酸-丙酮酸最小量运动试验,多项比值异常,具体结果详见表1。影像学检查:患者自2006—2014年多次行头颅CT及MRI检查,其中本次入院行头颅CT及MRI示小脑及脑干萎缩,双侧基底节及皮质下多发缺血变性灶及软化灶(图1)。眼底照相示:视盘边界清,色暗黄,血管走行僵直呈一致性狭窄;眼底色泽污秽,可见骨细胞样色素沉着(图2),提示视网膜色素变性。
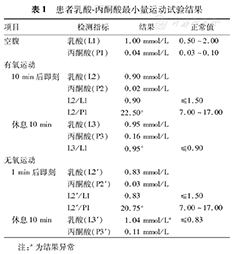
患者乳酸-丙酮酸最小量运动试验结果
患者乳酸-丙酮酸最小量运动试验结果
| 项目 | 检测指标 | 结果 | 正常值 | |
|---|---|---|---|---|
| 空腹 | 乳酸(L1) | 1.00 mmol/L | 0.50~2.00 | |
| 丙酮酸(P1) | 0.04 mmol/L | 0.03~0.10 | ||
| 有氧运动 | ||||
| 10 min后即刻 | 乳酸(L2) | 0.90 mmol/L | ||
| 丙酮酸(P2) | 0.02 mmol/L | |||
| L2/L1 | 0.90 | ≤1.50 | ||
| L2/P1 | 22.50a | 7.00~17.00 | ||
| 休息10 min | 乳酸(L3) | 0.95 mmol/L | ||
| 丙酮酸(P3) | 0.16 mmol/L | |||
| L3/L1 | 0.95a | ≤0.90 | ||
| 无氧运动 | ||||
| 1 min后即刻 | 乳酸(L2') | 0.83 mmol/L | ||
| 丙酮酸(P2') | 0.03 mmol/L | |||
| L2'/L1 | 0.83 | ≤1.50 | ||
| L2'/P1 | 20.75a | 7.00~17.00 | ||
| 休息10 min | 乳酸(L3') | 1.04 mmol/La | ≤0.83 | |
| 丙酮酸(P3') | 0.11 mmol/L | |||
注:a为结果异常
神经电生理检查示:双侧胫神经踇展肌记录运动波幅明显下降,传导速度轻度下降,双正中神经、右尺神经、双腓总神经运动传导正常;双正中神经、右尺神经感觉传导波幅明显下降,传导速度正常,左尺神经、双腓浅神经、双腓肠神经感觉传导未引出肯定波形;针极肌电示左背侧第一骨间肌静息状态下可见中等量正锐波,余所检肌肉未见异常自发电位,双背侧第一骨间肌、右胫前肌、左侧胸锁乳突肌小力收缩运动单位电位波幅升高,时限增宽,大力募集减少,左三角肌小力收缩运动单位电位波幅升高,时限正常范围,可疑早募集。最终结论为:神经传导示周围神经病变,感觉神经受累重于运动神经,轴索变性;针极肌电图示双背侧第一骨间肌、右胫前肌及左胸锁乳突肌呈神经源性损害,左三角肌可疑肌源性损害。
左肱二头肌病理结果:HE染色可见肌纤维大小略不等,偶见长条及小角形纤维,改良Gomori三色染色未见不整红边纤维(ragged red fiber,RRF),偶见肌纤维肌膜下轻度红染,琥珀酸脱氢酶(SDH)染色偶见SDH反应轻度增强血管,余染色未见特异性改变(图3)。

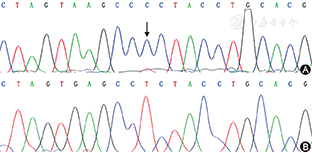
线粒体基因测序分析结果:提取患者骨骼肌DNA,行线粒体DNA全长测序,发现1个致病性突变位点,即mtDNA9185T>C,突变负荷约为80%(图4)。
讨论 NARP综合征是由线粒体基因突变导致的一种母系遗传的线粒体脑肌病,1990年由Holt等[1]首先报道,其临床表现主要表现为以下3个方面:①周围神经病或神经源性肌无力:主要表现为以感觉神经受累为主的周围神经病及近端肌无力伴病态疲劳;②共济失调:可能为首发症状,儿童期即可出现,表现为小脑性共济失调,头颅MRI可见小脑脑干萎缩;③视网膜色素变性:可为唯一的临床症状,最初表现为夜盲,随着疾病的进展出现视野缺损,最终可导致视力丧失,眼底检查可发现视网膜存在骨细胞样色素沉着。除上述典型表现外,患者还可出现癫痫发作、智力下降、神经性耳聋、心脏传导阻滞等线粒体脑肌病表现。多数患者疾病进展缓慢,可在多年内保持稳定,感染可使患者病情突然恶化[2]。NARP患者的骨骼肌病理表现轻微[1],可表现为正常、轻度肌源或神经源性损害,部分患者可出现肌纤维内脂滴轻度增多,很少出现线粒体脑肌病中常见的RRF。本例患者以共济失调、视力障碍起病,逐渐出现认知功能下降、癫痫发作、四肢无力,眼底检查提示视网膜色素变性,符合NARP的临床特点。
NARP主要由编码ATP酶F0结构域第6亚基的ATP6基因发生突变导致线粒体ATP生成障碍而致病。目前最常见且研究最多的点突变为T8993G,对于该位点而言,不同的突变负荷可导致不同的临床表现,当突变负荷小于70%时,患者可不出现临床症状,当突变负荷在70%~90%之间时可表现为NARP,而大于90%时可表现为亚急性坏死性脑脊髓病(Leigh病)[3]。由于线粒体突变负荷具有组织异质性的特点[4],NARP与Leigh病叠加综合征亦有报道,因此NARP与Leigh病可能同属ATP6基因突变相关疾病谱[5],两者并无绝对的界限。与T8993G突变不同的是,T9185C突变相关的临床表现有较大的异质性且与突变负荷无明显相关性,其中多数患者符合Leigh病的临床特点[6,7,8,9,10],而部分患者的受累较局限,表现为脊髓小脑共济失调[11]、腓骨肌萎缩症2型[12]、周期性瘫痪[13]及下运动神经元综合征[14],该突变导致典型NARP的病例尚未见报道。本例患者的骨骼肌突变负荷相对较低,肌病症状较轻,但中枢和周围神经以及视网膜损害较重,推测上述神经组织可能存在较高的突变负荷。
总之,本例NARP患者的临床表现较为典型,mtDNA T9185C突变导致经典NARP首次在我国被详细报道,结合国外相关文献,其表型变异性的原因值得进一步深入研究。

























