
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
自身免疫性肝炎(autoimmune hepatitis, AIH)是一种由针对肝细胞的自身免疫反应所介导的肝脏实质炎性反应,以血清自身抗体阳性、高免疫球蛋白G和(或)γ-球蛋白血症、肝组织学上存在界面性肝炎为特点,如不治疗常可导致肝硬化、肝功能衰竭[1]。AIH的临床表现多样,一般表现为慢性、隐匿起病,但也可表现为急性发作,甚至引起急性肝功能衰竭。20世纪60至80年代,多项临床研究证实,免疫抑制剂治疗可显著改善AIH患者的生物化学指标及临床症状,甚至能逆转肝纤维化,从而显著改善患者预后和生存质量。随着自身抗体和肝活组织病理学检查的广泛开展,我国AIH患者检出率逐年增加。AIH已成为非病毒性肝病的重要组成部分,越来越受到我国消化和肝病学界专家和临床医师们的关注[2]。
为进一步规范我国AIH的诊断和治疗,中华医学会肝病学分会、中华医学会消化病学分会和中华医学会感染病学分会组织国内有关专家制订了《自身免疫性肝炎诊断和治疗共识(2015)》。本共识旨在帮助医师在AIH诊治工作中做出合理决策。临床医师应充分了解本病的临床特点和诊断要点,认真分析单个病例的具体病情,进而制订出全面合理的诊疗方案。
本共识采用推荐意见分级的评估、制定和评价(GRADE)系统,对循证医学证据的质量和推荐强度等级进行评估,见表1, 表2。

GRADE系统的循证医学证据质量及其定义
GRADE系统的循证医学证据质量及其定义
| 证据级别 | 定义 |
|---|---|
| 高质量(A) | 非常确信估计的效应值接近真实的效应值,进一步研究也不可能改变该估计效应值的可信度 |
| 中等质量(B) | 对估计的效应值确信程度中等,估计值有可能接近真实值,但仍存在两者不相同的可能性,进一步研究有可能改变该估计效应值的可信度 |
| 低质量(C) | 对估计的效应值的确信程度有限:估计值与真实值可能大不相同。进一步研究极有可能改变该估计效应值的可信度 |
| 极低质量(D) | 对估计的效应值几乎没有信心:估计值与真实值很可能完全不同。对效应值的任何估计都很不确定 |

GRADE系统的推荐强度等级及其定义
GRADE系统的推荐强度等级及其定义
| 推荐强度 | 定义 |
|---|---|
| 强推荐(1级) | 明确显示干预措施利大于弊或者弊大于利 |
| 弱推荐(2级) | 利弊不确定或无论质量高低的证据均显示利弊相当 |
注:在形成推荐意见时,不仅考虑到证据的质量,还要权衡干预措施的利弊平衡与负担、患者偏好和价值观的不确定性或可变性,以及资源的合理利用、推荐措施的公平性与可实施性等
女性易患AIH,男女比例约为1∶4。AIH呈全球性分布,可发生于任何年龄段,但大部分患者年龄>40岁。最近,我国开展的一项全国范围内的回顾性调查(入选患者年龄>14岁)发现,AIH的峰值年龄为51岁(范围14~77岁),89%为女性患者。北欧白人的平均年发病率为(1.07~1.9)/10万,患病率为16.9/10万,而阿拉斯加居民的患病率可高达42.9/10万[3]。丹麦一项全国范围流行病学调查结果显示,年发病率为1.68/10万,且AIH的发病率有逐年增高趋势[4]。亚太地区的患病率介于(4~24.5)/10万,年发病率为(0.67~2)/10万[2]。目前,我国尚缺乏AIH流行病学的研究数据。
AIH临床表现多样,大多数AIH患者起病隐匿,一般表现为慢性肝病。最常见的症状包括嗜睡、乏力、全身不适等。体格检查时可发现肝大、脾大、腹水等体征,偶见周围性水肿。约1/3患者诊断时已存在肝硬化表现,少数患者以食管胃底静脉曲张破裂出血引起的呕血、黑便为首发症状。少部分患者可伴发热症状。10%~20%的患者无明显症状,仅在体检时意外发现血清转氨酶水平升高。这些无症状患者进展至肝硬化的危险性与有症状患者相近。AIH可在女性妊娠期或产后首次发病,早期诊断和及时处理对于母婴安全非常重要[5]。
约25%的AIH患者表现为急性发作,甚至可进展至急性肝功能衰竭。部分患者AIH病情可呈波动性或间歇性发作,临床和生物化学异常可自行缓解,甚至在一段时间内完全恢复,但之后又会复燃。这种情况需引起高度重视,因为这些患者的肝组织学仍表现为慢性炎性反应的持续活动,不及时处理可进展至肝纤维化。
AIH常合并其他器官或系统性自身免疫性疾病如:桥本甲状腺炎(10%~23%)、糖尿病(7%~9%)、炎症性肠病(2%~8%)、类风湿性关节炎(2%~5%)、干燥综合征(1%~4%)、银屑病(3%)和系统性红斑狼疮(1%~2%)等[3]。AIH和其他自身免疫性疾病,如系统性红斑狼疮,均为独立的疾病类型,若同时存在可按主要疾病类型处理,糖皮质激素剂量以能控制疾病活动为主。
AIH的典型血清生物化学指标异常主要表现为肝细胞损伤型改变,血清AST和ALT活性升高,而血清碱性磷酸酶(ALP)和γ-GT水平正常或轻微升高。应该注意的是,血清转氨酶水平并不能精确地反映肝内炎性反应情况。血清转氨酶水平正常或轻度异常不一定等同于肝内轻微或非活动性疾病,也不能完全排除AIH诊断。病情严重或急性发作时血清TBil水平可显著升高。
①血清免疫球蛋白。免疫球蛋白G(IgG)和(或)γ-球蛋白升高是AIH特征性的血清免疫学改变之一。血清IgG水平可反映肝内炎性反应活动程度,经免疫抑制剂治疗后可逐渐恢复正常。因此,该项指标不仅有助于AIH的诊断,而且对检测治疗应答具有重要的参考价值,在初诊和治疗随访过程中应常规检测。由于血清IgG水平的正常范围较宽,部分(5%~10%)患者基础IgG水平较低,疾病活动时即使IgG水平有所升高,但仍处于正常范围内,而治疗后检测可见到IgG水平的明显下降[6]。IgG4是IgG的4个亚群之一,占正常人血清IgG的5%,其抗原亲和力差,也缺乏结合C1q补体的能力。血清IgG4高于正常参考值(≥1 350 mg/L)可作为IgG4相关疾病、包括IgG4相关硬化性胆管炎的血清学诊断标准之一,但在AIH中的价值尚不明确[7]。AIH患者中血清IgM水平一般正常,血清IgA水平偶见升高,见图1。②自身抗体与分型。大多数AIH患者血清中存在一种或多种高效价的自身抗体,但这些自身抗体大多缺乏疾病特异性。病程中抗体效价可发生波动,但自身抗体效价并不能可靠地反映疾病的严重程度[8]。AIH可根据自身抗体的不同分为两型:抗核抗体(antinuclear antibodies, ANA)和(或)抗平滑肌抗体(anti-smooth muscle antibodies, ASMA),或抗可溶性肝抗原/肝胰抗原抗体(anti-soluble liver antigen/liver pancreas antigen,抗-SLA/LP)阳性者为1型AIH;抗肝肾微粒体抗体-1型(anti-liver kidney microsome-1,抗LKM-1)和(或)抗肝细胞溶质抗原-1型(anti liver cytosol-1,抗LC-1)阳性者为2型AIH,见图1。

注:AMA为抗线粒体抗体;抗LKM-1为抗肝肾微拉体抗体-1型;抗LC-1为抗肝细胞溶质抗原-1型;ANA为抗核抗体;ASMA为抗平滑肌抗体;SLA/LP为可溶性肝抗原/肝胰抗原抗体;↑为升高;↑↑为明显升高
临床上,70%~80%的AIH患者呈ANA阳性,20%~30%呈ASMA阳性(国内报道阳性率多低于欧美国家),ANA和(或)ASMA阳性者为80%~90%。ANA和ASMA为非器官组织特异性自身抗体,在高效价阳性时支持AIH诊断,低效价阳性可见于各种肝病甚至正常人。间接免疫荧光法可见ANA在细胞或组织切片上的荧光模式(核型)以核均质型略多见,也常见到多核点型、细颗粒型及两种或多种模式混合型。目前尚未发现不同荧光模式在AIH中的临床意义。ASMA与多种细胞骨架成分包括微丝、微管和中间丝反应。ASMA的主要靶抗原是微丝中的肌动蛋白,后者又可分为G-肌动蛋白和F-肌动蛋白。高效价抗F-肌动蛋白诊断AIH的特异度较高。研究显示,ASMA(>1∶80)和抗肌动蛋白抗体(>1∶40)与1型AIH患者的血清生物化学指标和组织学疾病活动度有关,并预示治疗失败概率较高[9]。
ANA是一组自身抗体的总称,检测方法有多种,不同方法所报告结果可能存在很大差异。目前,ANA和ASMA检测推荐间接免疫荧光法作为首选方法,检测结果推荐以效价值表示。在我国,自身抗体检测主要有两种稀释体系,不同体系之间的结果不具有固定的对应关系。ANA和ASMA效价越高,与自身免疫性疾病的相关性越大。临床高度疑似自身免疫性肝病的患者,建议进一步检测ANA中的特异性抗体(如dsDNA、SSA/SSB、gp210、sp100等)以帮助临床诊断与鉴别诊断。
抗-SLA对AIH具有高度诊断特异度,国内外报道其特异度均接近100%,但检出率较低,我国多中心自身免疫性肝病回顾性调查结果显示,仅6%(16/248)的患者呈抗-SLA阳性,明显低于欧美常见报道(30%左右)。抗-SLA阳性者往往同时存在ANA。SLA可能具有一定程度的致病性,有报道认为该抗体阳性与炎性反应较重、进展较快、易复发等特性有关[10]。我国研究结果显示,AIH患者对SLA抗原表位存在特异性T淋巴细胞免疫应答,并与肝细胞损伤的严重程度相关[11]。因此,有学者建议将抗-SLA阳性者归为3型AIH,但目前国际学术界尚有争议。
少数AIH患者(3%~4%)呈抗LKM-1和(或)抗LC-1阳性,可诊断为2型AIH。抗LKM-1阳性患者常呈ANA和SMA阴性,因此抗LKM-1的检测可避免漏诊AIH。抗LKM-1的靶抗原为细胞色素P450 2D6,已在AIH患者肝内检测到针对该自身抗原的CD4+和CD8+ T淋巴细胞的存在。LC-1所识别的靶抗原是亚氨甲基转移酶-环化脱氨酶。在10%的2型AIH患者中,LC-1是唯一可检测到的自身抗体,且抗LC-1与AIH的疾病活动度和进展有关。
此外,对那些常规自身抗体阴性却仍疑诊AIH的患者,建议检测其他自身抗体如:非典型核周型抗中性粒细胞胞质抗体(atypical perinuclear anti-neutrophilic cytoplasmic antibodies, pANCA)和抗去唾液酸糖蛋白受体抗体(antibodies against asialoglycoprotein receptor, ASGPR)等。
肝组织学检查对AIH的诊断和治疗非常重要。肝组织学检查的临床意义包括:可明确诊断、精确评价肝病分级和分期;多数自身抗体阴性患者(10%~20%)的血清IgG和(或)γ-球蛋白水平升高不明显,肝组织学检查可能是确诊的唯一依据;有助于与其他肝病(如药物性肝损伤、Wilson病等)鉴别,明确有无与其他自身免疫性肝病如:原发性胆汁性胆管炎(primary biliary cholangitis, PBC)[曾用名:原发性胆汁性肝硬化(primary biliary cirrhosis, PBC)]和原发性硬化性胆管炎(primary sclerosing cholangitis, PSC)的重叠存在;可协助判断合适的停药时机。肝组织学仍有轻度界面炎的患者停用免疫抑制剂后80%以上会复发[12]。因此,建议所有拟诊AIH的患者尽可能行肝组织学检查以明确诊断。AIH特征性肝组织学表现包括界面性肝炎、淋巴浆细胞浸润、肝细胞玫瑰花环样改变、淋巴细胞穿入现象和小叶中央坏死等。
由于门管区炎性反应导致与门管区或纤维间隔相邻的肝细胞坏死,称为界面性肝炎(interface hepatitis),表现为界面处肝细胞呈单个或小簇状坏死、脱落,导致小叶界面呈"虫蛀"状改变,旧称碎屑样坏死。炎性细胞沿破坏的界面向小叶内延伸,严重时可形成桥接坏死。按界面破坏范围和浸润深度,可分为轻、中、重度界面性肝炎,轻度:局部或少数门管区破坏;中度:<50%的门管区或纤维间隔破坏;重度:>50%的门管区或纤维间隔破坏。中、重度界面性肝炎支持AIH的诊断。界面性肝炎是AIH的组织学特征之一,但特异度并不高,轻度界面性肝炎也可存在于其他慢性肝病如病毒性肝炎、药物性肝损伤、Wilson病等。
AIH患者肝组织门管区及其周围浸润的炎性细胞主要为淋巴细胞和浆细胞。浆细胞浸润是AIH另一特征性组织学改变,主要见于门管区和界面处,有时也可出现在小叶内。但浆细胞缺如并不能排除AIH的诊断,约1/3的AIH患者可表现为浆细胞稀少甚至缺如。AIH中的浆细胞主要呈胞质IgG阳性,少量为IgM阳性(PBC中浆细胞以IgM为主)。
肝细胞呈"玫瑰花环"样改变是指由数个水样变性的肝细胞形成的假腺样结构,中心有时可见扩张的毛细胆管,形似玫瑰花环,周围可见淋巴细胞包绕,一般见于界面炎周围。
穿入现象是指淋巴细胞进入肝细胞胞质的组织学表现,多见于活动性界面炎区域。我国研究结果表明,65%的AIH患者可见穿入现象,显著高于其他慢性肝病患者,并与AIH肝内炎性反应和纤维化程度相关。穿入的淋巴细胞主要为CD8+T淋巴细胞,可导致肝细胞凋亡[13]。
研究结果显示,17.5%的AIH患者在肝活组织检查中可出现小叶中央(第三区)坏死,可能是AIH急性发作的表现之一。它可以单独出现,也可伴随界面性肝炎和较重的门管区炎性反应。患者往往伴有高TBil血症,及时的免疫抑制剂治疗缓解后小叶中央坏死可完全消失。
临床上如遇到不明原因肝功能异常和(或)肝硬化的任何年龄、性别患者,均应考虑AIH的可能。国际自身免疫性肝炎小组(International Autoimmune Hepatitis Group, IAIHG)于1993制定了AIH描述性诊断标准和诊断积分系统,并于1999年进行了更新,见表3, 表4[14]。1999年更新的积分系统根据患者是否已接受糖皮质激素治疗,分为治疗前和治疗后评分。治疗前评分中临床特征占7分,实验室检查占14分,肝组织病理学占5分,确诊需评分≥16分,10~15分为可能诊断,低于10分可排除AIH诊断。治疗后评分除上述项目外,还包括患者对治疗反应(完全或复发)的评分,确诊需评分≥18分,12~17分为可能诊断。该系统主要适用于具有复杂表现患者的诊断,多用于临床研究。对包含983例患者的6项研究进行Meta分析后发现,其诊断准确度为89.8%。该系统在鉴别"确诊性AIH"和胆汁淤积性肝病(PBC和PSC)时有较好的特异度。

自身免疫性肝炎描述性诊断标准[14]
自身免疫性肝炎描述性诊断标准[14]
| 特征 | 明确 | 可能 |
|---|---|---|
| 肝组织学 | 中度或重度界面性肝炎、小叶性肝炎或中央区-汇管区桥接坏死,但无胆管病变、明确的肉芽肿或其他提示特定病因的组织学特点 | 同"明确"栏 |
| 血清生物化学检查 | 血清转氨酶不同程度的升高,特别是(但不排除性的)血清碱性磷酸酶升高不明显。血清α1-抗胰蛋白酶、血清铜和铜蓝蛋白浓度正常 | 同"明确"栏,但如果Wilson病被排除后,可包括血清铜和铜蓝蛋白浓度异常的患者 |
| 血清免疫球蛋白 | 血清γ-球蛋白或IgG水平超过正常上限的1.5倍 | 血清γ-球蛋白或IgG水平超过正常上限的任何升高 |
| 血清抗体 | 血清ANA、ASMA或抗LKM-1抗体效价>1∶80。较低的效价(特别是抗LKM-1)在儿童中也有价值 | 同"明确"栏,抗体效价为1∶40或以上。这些血清抗体阴性,但也包括其他特定的抗体阳性者 |
| 病毒标志物 | 目前感染甲型、乙型或丙型肝炎的病毒标志物阴性 | 同"明确"栏 |
| 其他致病因素 | 平均乙醇消耗量<25 g/d。最近无已知的肝毒性药物服用史 | 乙醇消耗量<50 g/d,最近无肝毒性药物服用史。如果有确切的证据表明在戒酒和停用药物后持续存在肝损害,消耗较多乙醇的患者或最近服用肝毒性药物的患者也可包括在内 |
注:ANA为抗核抗体;ASMA为抗平滑肌抗体;抗LKM-1为抗肝肾微粒体抗体-1型

AIH综合诊断积分系统(1999年)[14]
AIH综合诊断积分系统(1999年)[14]
| 参数/临床特征 | 计分 | 参数/临床特征 | 计分 | ||||
|---|---|---|---|---|---|---|---|
| 女性 | +2 | 药物史 | |||||
| ALP(正常上限倍数)/AST(或ALT)(正常上限倍数)的比值 | 阳性 | -4 | |||||
| 阴性 | +1 | ||||||
| 平均乙醇摄入量 | |||||||
| <1.5 | +2 | <25 g/d | +2 | ||||
| 1.5~3.0 | 0 | >60 g/d | -2 | ||||
| >3.0 | -2 | 肝脏组织学检查 | |||||
| 血清γ-球蛋白或IgG与正常值的比值 | 界面性肝炎 | +3 | |||||
| 主要为淋巴-浆细胞浸润 | +1 | ||||||
| >2.0 | +3 | 肝细胞呈玫瑰花环样改变 | +1 | ||||
| 1.5~2.0 | +2 | ||||||
| 1.0~1.5 | +1 | 无上述表现 | -5 | ||||
| <1.0 | 0 | 胆管改变 | -3 | ||||
| ANA, SMA或LKM-1效价 | 其他改变 | -3 | |||||
| 其他免疫性疾病 | +2 | ||||||
| >1∶80 | +3 | 其他可用的参数 | |||||
| 1∶80 | +2 | 其他特异性自身抗体(SLA/LP、LC-1、ASGPR、pANCA)阳性 | +2 | ||||
| 1∶40 | +1 | ||||||
| <1∶40 | 0 | ||||||
| AMA阳性 | -4 | HLA-DR3或DR4 | +1 | ||||
| 肝炎病毒标志物 | 对治疗的反应 | ||||||
| 阳性 | -3 | 完全 | +2 | ||||
| 阴性 | +3 | 复发 | +3 | ||||
| 总积分的解释 | 总积分的解释 | ||||||
| 治疗前: | 治疗后: | ||||||
| 明确的AIH | ≥16 | 明确的AIH | ≥18 | ||||
| 可能的AIH | 10~15 | 可能的AIH | 12~17 | ||||
注:AIH为自身免疫性肝炎;ALP为碱性磷酸酶;AST为天冬氨酸转氨酶;ALT为丙氨酸转氨酶;ANA为抗核抗体;ASMA为抗平滑肌抗体;抗LKM-1为抗肝肾微粒体抗体-1型;AMA为抗线粒体抗体;SLA/LP为抗可溶性肝抗原/肝胰抗原抗体;抗LC-1为抗肝细胞溶质抗原-1型;ASGPR为抗去唾液酸糖蛋白受体抗体;pANCA为非典型核周型抗中性粒细胞胞质抗体;HLA为人类白细胞抗原
虽然综合诊断积分系统诊断AIH时具有较高的灵敏度和特异度,但较复杂,难以在临床实践中全面推广。有鉴于此,2008年IAIHG提出了AIH简化诊断积分系统,见表5。简化诊断积分系统分为自身抗体、血清IgG水平、肝组织学改变和排除病毒性肝炎等四个部分,每个组分最高计2分,共计8分。积分6分者为"可能"的AIH;积分≥7分者可确诊AIH[15]。我国一项总数为405例慢性肝病患者(其中1型AIH患者127例)的多中心临床研究结果显示,简化积分系统确诊AIH的灵敏度为90%,特异度为95%,可较好地应用于临床诊断[16]。综合几项规模较大的验证结果发现,AIH简化积分系统在诊断"可能"的AIH(即6分)时的中位灵敏度为91%(范围65%~95%),中位特异度为94%(范围90%~98%);而诊断"明确"的AIH(即≥7分)时,其中位灵敏度和特异度分别是75.5%(范围15%~87%)、100%(范围100%)。但简化积分系统容易漏诊部分不典型患者,如自身抗体效价低或阴性和(或)血清IgG水平较低甚至正常的患者。因此,对于疑似患者而简化诊断积分不能确诊的患者,建议再以综合诊断积分系统进行综合评估以免漏诊[16]。

国际自身免疫性肝炎小组的AIH简化诊断标准[15]
国际自身免疫性肝炎小组的AIH简化诊断标准[15]
| 变量 | 标准 | 分值 | 备注 |
|---|---|---|---|
| ANA或ASMA | ≥1∶40 | 1分 | 相当于我国常用的ANA 1∶100的最低效价 |
| ANA或ASMA、抗LKM-1、抗SLA阳性 | ≥1∶80 | 2分 | 多项同时出现时最多2分 |
| ≥1∶40 | |||
| 阳性 | |||
| >正常值上限 | 1分 | ||
| >1.10倍正常值上限 | 2分 | ||
| 肝组织学 | 符合AIH | 1分 | 界面性肝炎、汇管区和小叶内淋巴-浆细胞浸润、肝细胞玫瑰样花环以及穿入现象被认为是特征性肝组织学改变,4项中具备3项为典型表现 |
| 典型AIH表现 | 2分 | ||
| 排除病毒性肝炎 | 是 | 2分 | |
| =6分:AIH可能 | |||
| ≥7分:确诊AIH |
注:AIH为自身免疫性肝炎;ANA为抗核抗体;ASMA为抗平滑肌抗体;抗LKM-1为抗肝肾微粒体抗体-1型;抗-SLA为抗肝可溶性抗原抗体
ANA和ASMA等自身抗体缺乏疾病特异性,低效价的自身抗体也可见于其他多种肝内、外疾病,如病毒性肝炎、非酒精性脂肪性肝病、Wilson病等肝病以及系统性红斑狼疮、类风湿性关节炎等自身免疫性疾病。因此,需进行仔细的鉴别诊断,见表6。

自身免疫性肝炎的鉴别诊断
自身免疫性肝炎的鉴别诊断
| 疾病 | 临床表现和实验室检查 | 病理学表现 |
|---|---|---|
| HCV感染 | 血清ANA可低效价阳性或抗LKM-1阳性,IgG水平轻度升高;抗-HCV和HCV RNA阳性 | 肝细胞脂肪变性、淋巴滤泡形成、肉芽肿形成 |
| 药物性肝损伤 | 药物史明确,停用药物后好转;血清转氨酶水平升高和(或)胆汁淤积表现 | 汇管区中性粒细胞和酸性粒细胞浸润、肝细胞大泡脂肪变性、肝细胞胆汁淤积,纤维化程度一般较轻(低于S2) |
| 非酒精性脂肪性肝病 | 1/3患者血清ANA可低效价阳性,血清转氨酶轻度升高,胰岛素抵抗表现 | 肝细胞呈大泡脂肪变性、肝窦纤维化、汇管区炎性反应较轻 |
| Wilson病 | 血清ANA可阳性,血清铜蓝蛋白低,24 h尿铜升高,可有角膜色素环(K-F环)阳性 | 存在肝细胞脂肪变性、空泡状核形成、汇管区炎性反应,可伴界面炎,可有大量铜沉着 |
注:HCV为丙型肝炎病毒;ANA为抗核抗体;抗-LKM-1为抗肝肾微粒体抗体-1型
推荐意见1: AIH主要表现为慢性肝炎、肝硬化,也可表现为急性发作,甚至急性肝功能衰竭。因此,原因不明的肝功能异常患者均应考虑存在AIH的可能(1B)。
推荐意见2:拟诊AIH时应检测肝病相关自身抗体,并可根据自身抗体将AIH分为两型:1型AIH呈ANA、ASMA或抗-SLA/LP阳性,2型AIH呈抗LKM-1和(或)抗LC-1阳性(1B)。
推荐意见3:拟诊AIH时应检测血清IgG和(或)γ-球蛋白水平,血清免疫球蛋白水平对诊断和观察治疗应答有重要价值(1B)。
推荐意见4:应尽可能对所有拟诊AIH的患者进行肝组织学检查以明确诊断。AIH特征性肝组织学表现包括界面性肝炎、淋巴-浆细胞浸润、肝细胞玫瑰花环样改变和淋巴细胞穿入现象等(1B)。
推荐意见5: AIH患者常并发其他器官或系统性自身免疫性疾病(1C)。
推荐意见6: AIH的诊断应结合临床症状与体征、血清生物化学、免疫学异常、血清自身抗体以及肝脏组织学等进行综合诊断,并排除其他可能病因(1A)。
推荐意见7:简化积分系统可用于我国AIH患者的临床诊断,具有较高的灵敏度和特异度。但遇到临床表现、血清生物化学指标和免疫学或肝组织学不典型的病例时,可使用综合评分系统进行评估(1B)。
推荐意见8:诊断AIH时需注意与药物性肝损伤、慢性HCV感染、Wilson病和非酒精性脂肪性肝炎等肝脏疾病进行鉴别,合并胆汁淤积表现时需与PBC、PSC和IgG4相关硬化性胆管炎等鉴别(1A)。
AIH治疗的总体目标是获得肝组织学缓解、防止肝纤维化的发展和肝功能衰竭的发生,延长患者的生存期和提高患者的生存质量。临床上可行的治疗目标是获得完全生物化学指标缓解,即血清转氨酶(ALT/AST)和IgG水平均恢复正常[17]。研究结果表明,肝组织学完全缓解者[Ishak组织学活动指数(histological activity index, HAI)<3]较之未获得组织学完全缓解者(HAI≥4)肝纤维化逆转率较高(60%与32%,P<0.01),长期生存期也显著延长[18]。因此,肝组织学缓解可能是治疗的重要目标。
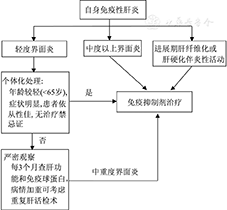
所有活动性AIH患者均应接受免疫抑制剂治疗,并可根据疾病活动度调整治疗方案和药物剂量。
1.中度以上炎性反应活动的AIH患者[血清转氨酶水平>3×正常值上限(ULN)、IgG>1.5×ULN],急性[ALT和(或)AST>10×ULN]甚至重症[伴出、凝血异常:国际标准化比率(INR)>1.5]应及时启动免疫抑制剂治疗,以免出现急性肝功能衰竭。
2.对于轻微炎性反应活动(血清转氨酶水平<3×ULN、IgG<1.5×ULN)的老年(>65岁)患者,需平衡免疫抑制剂治疗的益处和风险作个体化处理。暂不启动免疫抑制剂治疗者需严密观察,如患者出现明显的临床症状,或出现明显炎性反应活动可进行治疗。
3.从肝组织学角度判断,存在中度以上界面性肝炎是治疗的重要指征,见图2。桥接性坏死、多小叶坏死或塌陷性坏死、中央静脉周围炎等特点,提示急性或重症AIH,需及时启动免疫抑制剂治疗。轻度界面炎患者可视年龄而区别对待。轻度界面性肝炎的老年患者可严密观察、暂缓用药,特别是存在免疫抑制剂禁忌证者。而存在轻度界面炎的年轻患者仍有进展至肝硬化的风险,可酌情启动免疫抑制剂治疗。对非活动性肝硬化AIH患者则无需免疫抑制剂治疗,但应长期密切随访(如每隔3~6个月随访1次)。
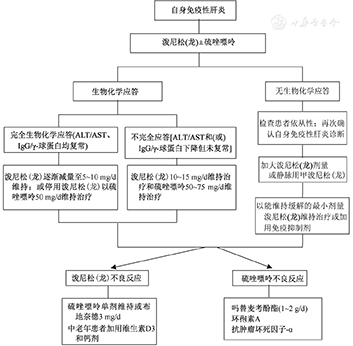
AIH患者一般优先推荐泼尼松(龙)和硫唑嘌呤联合治疗方案,联合治疗可显著减少泼尼松(龙)剂量及其不良反应。泼尼松(龙)可快速诱导症状缓解、血清转氨酶和IgG水平的复常,用于诱导缓解,而硫唑嘌呤需6~8周才能发挥最佳免疫抑制效果,多用于维持缓解。最近,欧洲肝病学会AIH指南建议在使用泼尼松(龙)2周出现显著生物化学应答后再加用硫唑嘌呤,也是一个值得借鉴的治疗策略[19]。联合治疗特别适用于同时存在下述情况的AIH患者,如绝经后妇女、骨质疏松、脆性糖尿病、肥胖、痤疮、情绪不稳及高血压患者。基于随机对照试验的Meta分析研究结果表明,泼尼松(龙)单药治疗和联合治疗在初治和复发的诱导缓解中均有效,而维持治疗中联合治疗或硫唑嘌呤单药治疗组的疗效优于泼尼松(龙)单药治疗[20]。泼尼松(龙)初始剂量为30~40 mg/d,并于4周内逐渐减量至10~15 mg/d;硫唑嘌呤以50 mg/d的剂量维持治疗。诱导缓解治疗一般推荐如下用药方案:泼尼松(龙)30 mg/d 1周、20 mg/d 2周、15 mg/d 4周,泼尼松(龙)剂量低于15 mg/d时,建议以2.5 mg/d的幅度渐减至维持剂量(5~10 mg/d);维持治疗阶段甚至可将泼尼松(龙)完全停用,仅以硫唑嘌呤50 mg/d单药维持。需要强调的是,糖皮质激素的减量应遵循个体化原则,可根据血清生物化学指标和IgG水平改善情况进行适当调整,如患者改善明显可较快减量,而疗效不明显时可在原剂量上维持2~4周。伴发黄疸的AIH患者可先以糖皮质激素改善病情,待TBil显著下降后再考虑加用硫唑嘌呤联合治疗。
泼尼松(龙)单药治疗时初始剂量一般选择40~60 mg/d,并于4周内逐渐减量至15~20 mg/d。初始剂量可结合患者症状、血清转氨酶和IgG水平,特别是肝组织学炎性反应程度进行合理选择。单药治疗适用于合并血细胞减少、巯基嘌呤甲基转移酶(thiopurine methyltransferase, TPMT)功能缺陷、妊娠或拟妊娠、并发恶性肿瘤的AIH患者。已有肝硬化表现者多选择泼尼松(龙)单药治疗并酌情减少药物剂量。AIH"可能"诊断患者也可以单剂泼尼松(龙)进行试验性治疗。泼尼松可在肝脏代谢为泼尼松龙后发挥作用,除非肝功能严重受损,两者作用相似。泼尼松龙可等剂量替代泼尼松,而4 mg的甲基泼尼松龙相当于5 mg泼尼松(龙)。
布地奈德(budesonide)是第二代糖皮质激素,其在肝脏的首过清除率较高(约90%),6-OH-布地奈德与糖皮质激素受体的亲和性高,抗炎疗效相当于泼尼松(龙)的5倍,而其代谢产物(16-OH-泼尼松龙)无糖皮质激素活性。因此,布地奈德作用的主要部位为肠道和肝脏,而全身不良反应较少。来自欧州的多中心临床研究结果表明,布地奈德和硫唑嘌呤联合治疗方案较传统联合治疗方案能更快诱导缓解,而糖皮质激素相关不良反应显著减轻,可作为AIH的一线治疗方案[21]。目前多用于需长期应用泼尼松(龙)维持治疗的AIH患者,以期减少糖皮质激素的不良反应。由于布地奈德与泼尼松一样作用于激素受体,因此,不推荐用于传统激素无应答的患者。在肝硬化门静脉侧支循环开放患者中,布地奈德可通过侧支循环直接进入体循环而失去首过效应的优势,同时还可能有增加门静脉血栓形成的风险。因此,布地奈德不宜在肝硬化患者中应用。
对标准治疗无效或不能耐受标准治疗不良反应的患者,可以选择二线治疗方案,目前已有应用吗替麦考酚酯(MMF)、环孢素A、他克莫司、6-巯基嘌呤、甲氨蝶呤、抗肿瘤坏死因子α等治疗难治性AIH的报道。MMF是在标准治疗效果不佳患者中应用最多的替代免疫抑制剂。泼尼松联合MMF作为AIH的一线治疗,可使88%的患者出现完全生物化学应答(即血清生物化学指标和血清IgG水平恢复正常),而且生物化学应答往往在治疗开始后的3个月内。12%的患者出现部分生物化学应答[22]。临床上,MMF在不能耐受硫唑嘌呤治疗的患者具有补救治疗作用,而对硫唑嘌呤无应答的患者MMF的疗效也较差。另外,在胆汁淤积性AIH患者中如糖皮质激素疗效欠佳,也可考虑加用小剂量MMF治疗,以避免硫唑嘌呤诱导胆汁淤积的不良反应。
应答不完全定义为经2~3年治疗后,临床表现、实验室指标[血清转氨酶、TBil、IgG和(或)γ-球蛋白]和肝组织学等改善,但未完全恢复正常[17]。免疫抑制剂治疗应答不佳或无应答者应首先考虑AIH诊断是否有误和患者对治疗的依从性如何。少数AIH患者确实显示对免疫抑制剂治疗应答不佳或应答不完全,部分患者可能在激素减量过程中或在维持治疗过程中出现反跳。该类患者可酌情短期(1周)给予大剂量甲泼尼龙(40~60 mg/d)静脉滴注,病情缓解后改为口服泼尼松龙治疗(30~40 mg/d),适当放缓减量速度,并加以免疫抑制剂维持治疗。泼尼松龙和硫唑嘌呤联合治疗2年仍未达到缓解的患者,建议继续用泼尼松龙(5~10 mg/d)+大剂量硫唑嘌呤[最高达2 mg/(kg·d)],12~18个月后复查肝活组织病理学检查。对于已接受至少36个月连续治疗但临床、实验室和组织学的改善未达到治疗终点的不完全应答患者,建议将泼尼松或硫唑嘌呤调整至适合剂量以长期维持治疗,使此类患者处于无症状、实验室指标稳定的状态。
免疫抑制剂治疗一般应维持3年以上,或获得生物化学缓解后至少2年以上。除完全生物化学应答外,停用免疫抑制剂的指征包括肝内组织学恢复正常、无任何炎性反应活动表现,因为即使轻度界面性肝炎的存在也预示着停药后复发的可能。复发可定义为血清转氨酶水平>3×ULN,伴血清IgG和(或)γ-球蛋白水平不同程度的升高[14,17,23]。停药后复发是AIH的临床特点之一,临床缓解至少2年的患者在停药1年后59%的患者需要重新治疗,2年后为73%,3年后高达81%;复发的危险因素包括先前需使用联合治疗方案才能获得生物化学缓解者、并发自身免疫性疾病和年龄较轻者[24]。以单剂免疫抑制剂治疗即可获得长期完全生物化学缓解至少2年以上的患者,获得持续缓解的可能性较高。虽然均在正常范围内,较高的血清ALT和IgG水平仍与复发相关。所有持续缓解的患者在停药时的ALT水平低于ULN的一半,而IgG水平低于12 g/L[25]。
停药后初次复发患者,建议再次以初始治疗的剂量给予泼尼松(龙)和硫唑嘌呤联合治疗,逐渐减量甚至停药,并以硫唑嘌呤(50~75 mg/d)维持治疗;而硫唑嘌呤不能耐受的患者可给予小剂量泼尼松(≤10 mg/d)或与MMF联合长期维持治疗。2次以上复发者建议以最小剂量长期维持治疗。
无论是单用泼尼松(龙)还是与硫唑嘌呤联合治疗,所有患者都必须监测相关的药物不良反应。约10%的患者可因药物不良反应而中断治疗。可选择该患者相对不良反应较小的免疫抑制剂进行治疗,如小剂量糖皮质激素、单剂硫唑嘌呤或二线免疫抑制剂MMF等,且必须尽量采用能控制疾病活动的最低剂量。
长期使用糖皮质激素可出现明显不良反应,其中除了常见的Cushing征(满月脸、痤疮、水牛背、向心性肥胖等)以外,糖皮质激素还可加重骨质疏松,导致脊柱压缩性骨折和股骨头缺血性坏死等骨病,并与2型糖尿病、白内障、高血压病、感染(包括已有的结核发生恶化)、精神疾病的发生有关。患者由于不能接受其外貌上的变化或肥胖,是造成治疗中断的最常见原因(占47%),其次为骨量减少造成的脊柱压缩(占27%)和脆性糖尿病(占20%)等。应尽量采用联合治疗方案,尽量减少糖皮质激素剂量,并最终过渡至硫唑嘌呤单药维持治疗方案。
需长期接受糖皮质激素治疗的AIH患者,建议治疗前做基线骨密度检测并每年监测随访。骨病的辅助治疗包括:坚持规律的负重锻炼、补充维生素D3和钙质,适时给予骨活性制剂如二磷酸盐治疗。
硫唑嘌呤最常见的不良反应是血细胞减少,可能与红细胞内TPMT活性低有关。因此,加用硫唑嘌呤的患者需严密监测血常规变化,特别是用药的前3个月。如发生血白细胞计数的快速下降或白细胞计数<3.5×109/L需紧急停用硫唑嘌呤。硫唑嘌呤的其他不良反应包括肝内胆汁淤积、静脉闭塞性疾病、胰腺炎、严重恶心呕吐、皮疹等。少于10%的患者在接受硫唑嘌呤(50 mg/d)时会出现上述不良反应,一般均可在减量或停用后改善。以下人群不推荐使用硫唑嘌呤:基础状态下已存在血细胞计数减少(白细胞计数<3.5×109/L或血小板计数<50×109 /L)、恶性肿瘤、已知TPMT功能缺陷等。硫唑嘌呤治疗前或治疗过程中出现血细胞减少的AIH患者,建议检测其血TPMT活性。
AIH的治疗策略见图3。
注:ALT为丙氨酸转氨酶;AST为天冬氨酸转氨酶
推荐意见9:治疗目标是获得生物化学缓解[血清转氨酶、IgG和(或)γ-球蛋白水平均恢复正常]和肝组织学缓解,防止疾病进展(1B)。
推荐意见10:中重度AIH、急性表现、活动性肝硬化等活动性AIH患者均建议行免疫抑制治疗(1A)。
推荐意见11:以肝组织学为依据,存在中、重度界面性肝炎的患者应行免疫抑制剂治疗。轻度界面性肝炎的年轻患者亦推荐行免疫抑制剂治疗,而存在轻度界面性肝炎的老年(>65岁)患者可暂不予免疫抑制剂治疗(1B)。
推荐意见12:对于无疾病活动或自动缓解期的AIH、非活动性肝硬化可暂不考虑行免疫抑制剂治疗,但应长期密切随访(如每隔3~6个月随访1次)(2C)。
推荐意见13:一般选择泼尼松(龙)和硫唑嘌呤联合治疗方案。推荐泼尼松(龙)初始剂量一般为30~40 mg/d,4~6周内逐渐减至15 mg/d,并以5~7.5 mg/d维持;硫唑嘌呤剂量为50 mg/d或1 mg/(kg·d),可尝试在维持治疗中完全停用泼尼松(龙)而以硫唑嘌呤单药维持治疗(1B)。
推荐意见14:选择泼尼松(龙)单药治疗方案时,推荐泼尼松(龙)初始剂量一般为40~60 mg/d,并于4~6周内逐渐减量至15~20 mg/d,以5~10 mg/d剂量维持治疗(1B)。
推荐意见15:提倡个体化治疗,应根据血清转氨酶和IgG恢复情况调整泼尼松(龙)的剂量(1B)。
推荐意见16:对于硫唑嘌呤应答但不能耐受者,可考虑在泼尼松(龙)的基础上加用MMF(0.5~1.0 g/d,分2次服用),但也应严密监测血常规变化(1B)。
推荐意见17:免疫抑制剂治疗一般应维持3年以上,或获得生物化学缓解后至少2年以上。建议停药前行肝组织学检查,肝内无炎性反应活动时方可考虑停药(1B)。
推荐意见18:停药后复发或维持治疗中反跳的AIH患者应以初始治疗相似的方案进行治疗,并推荐尽可能联合治疗并长期维持(1C)。
推荐意见19:需长期接受糖皮质激素治疗的AIH患者,建议治疗前行基线骨密度测定并每年监测随访,并适当补充维生素D和钙剂(1B)。
推荐意见20:在治疗前已存在血细胞计数减少者或肝硬化者,慎用硫唑嘌呤。硫唑嘌呤用药过程中也应注意检测全血细胞计数,防止骨髓抑制的发生。在有条件的情况下可检测TPMT基因型或活性以指导临床用药(1B)。
AIH患者如出现终末期肝病或急性肝功能衰竭等情况需考虑进行肝移植术。重症AIH可导致急性或亚急性肝功能衰竭,如短期(常常1周)的糖皮质激素治疗疗效不明显时,需及时与肝移植中心联系,以免失去紧急肝移植术机会。另一种情况是失代偿期肝硬化患者,其移植指征与其他病因导致的肝硬化相似,包括反复食管胃底静脉曲张出血、肝性脑病、顽固性腹水、自发性细菌性腹膜炎和肝肾综合征等并发症经内科处理疗效不佳,终末期肝病模型(MELD)>15分或Child-Pugh积分>10,或符合肝移植标准的肝细胞癌。选择恰当的时间进行肝移植术十分关键,应尽早做好肝移植术准备,而不是出现终末期肝病严重并发症再开始评估,因为慢加急性肝功能衰竭导致多器官衰竭常常使患者失去进行肝移植术的机会[26]。欧洲991例因AIH行肝移植术患者1年生存率为88%,移植物生存率为84%;5年患者生存率为80%,移植物生存率为72%,与PBC和PSC患者预后相似[26]。
20%的AIH患者在肝移植后会再次发病,中位诊断时间为肝移植术后26个月[27]。美国国家健康学会(NIH)肝移植数据库的人类白细胞抗原(HLA)匹配分析表明,HLA-DR位点的不匹配是复发性AIH的主要危险因素[28]。术前较高的血清IgG水平、移植肝的中、重度炎性反应与AIH复发有关,提示术前未能完全抑制疾病活动是复发的危险因素之一[29]。因此,AIH患者在肝移植术后的免疫抑制方案应兼顾抗排异反应和防止AIH复发。可在标准抗排异方案基础上以小剂量泼尼松(龙)长期维持,必要时加用硫唑嘌呤联合治疗[30]。
少数(6%~10%)非AIH患者在肝移植后出现类似AIH的血清学和组织学表现,称为"新发(de novo)"AIH[31]。成人"新发"AIH在HCV相关肝硬化经肝移植术后接受干扰素和病毒唑治疗后报道较多,但诊断相当困难[32]。我国学者曾报道1例"新发"AIH患者,该患者因HCV相关肝硬化在肝移植术后接受1年的聚乙二醇干扰素α-2b和病毒唑联合抗病毒治疗后出现AIH样特征。由于其血清IgG4显著增高,肝组织学上IgG4阳性浆细胞显著增加,因此将其定义为IgG4相关"新发"AIH[33]。
推荐意见21: AIH患者的肝移植指征包括:①终末期肝病经内科处理疗效不佳者;②急性肝功能衰竭经糖皮质激素治疗1周后病情无明显改善甚至恶化者(1B)。
推荐意见22:肝移植术后AIH复发的患者建议在抗排异治疗方案基础上加用泼尼松(龙)或硫唑嘌呤。因其他病因进行肝移植患者如出现AIH样生物化学和肝组织学表现,需考虑"新发"AIH(de novo AIH)的可能性(1C)。
AIH临床表现多样,大多表现为慢性肝病,但也可表现为急性发作、急性和慢性肝功能衰竭等。特殊人群如儿童、老年、孕妇也具有不同的临床特点。因此,我们需充分认识AIH的异质性和特殊性,并采取适当的治疗策略[34],见图3。
急性起病的AIH包含两种形式:慢性疾病基础上的急性恶化和真正意义上的无慢性疾病表现的急性AIH[19]。典型的AIH呈慢性病程,但高达25%的AIH患者可表现为急性起病,其中小部分可进展为自身免疫性急性肝功能衰竭[35]。急性起病的AIH通常表现为病程短(<30 d)且没有既往明确肝脏疾病史,临床症状明显(如黄疸、疲乏、发热、恶心、全身不适等),血清学明显异常(血清ALT>5×ULN,TBil>34.2 μmol/L)。小叶中央坏死是AIH急性起病的肝组织学特征,及时发现有助于早期诊断和干预[36]。在IAIHG提出的综合和简化诊断积分系统中,自身抗体和血清IgG水平是诊断AIH的两个重要因素,而急性起病的AIH常缺少这两个重要特征。简化诊断积分系统只能诊断出24%的急性AIH患者,而综合诊断积分系统则可诊断出40%的急性AIH患者[37]。美国急性肝功能衰竭协作网的报告表明,10%的急性肝功能衰竭患者是由AIH引起,另有30%的急性肝功能衰竭患者表现为"血清阴性",其中有一部分可能为AIH[38]。
来自日本的资料显示,早期、足量静脉使用甘草酸制剂可缓解急性发作性AIH的进展。因此,在启动特异性治疗前,甘草酸制剂可作为初始治疗,安全有效地用于不明原因的急性肝炎[18]。短期大剂量糖皮质激素(60 mg/d)治疗对36%~100%的急性起病AIH患者有效,治疗反应的差异与开始治疗是否及时有关。肝组织学上有小叶中央坏死的急性起病AIH患者倾向于对激素治疗反应良好,但有报道指出急性起病的AIH与普通AIH相比,更容易产生糖皮质激素抵抗[39]。急性起病的AIH患者对激素的反应与预后密切相关。使用糖皮质激素治疗2周内实验室检查指标未出现改善、且组织学上出现肝脏多小叶坏死的急性AIH患者,预后常极差。如果患者的高TBil血症未改善甚至加重,预示早期病死率极高,甚至达100%。MELD评分能有效评估风险及定量分析病情的改善或恶化。当MELD评分≥12分时,有97%的灵敏度和68%的特异度提示患者激素治疗可能失败[40]。因此,临床上可使用1~2周糖皮质激素来判断是否需继续激素治疗,同时必须进行MELD评分,判断是否需要肝移植术。
AIH患者可出现肝内胆汁淤积表现,约20%的胆汁淤积型(血清TBil≥40 μmol/L)AIH患者对糖皮质激素治疗无应答,并与病死率和肝移植率显著增高相关。治疗失败的最佳预测因素是糖皮质激素治疗1周前后MELD-Na和英国终末期肝病模型(UKELD)的变化。早期识别无应答者有助于及时增加免疫抑制剂剂量,防止临床恶化或及时转入肝移植中心[41]。熊去氧胆酸(UDCA)可有效缓解患者胆汁淤积表现,可联合使用。胆汁淤积型AIH患者在初期应避免使用硫唑嘌呤,以免加重胆汁淤积,可先使用大剂量糖皮质激素(如40~60 mg/d)缓解病情,可在血清TBil显著下降后再加用硫唑嘌呤联合治疗。
血清自身抗体是AIH的免疫学特征之一,约10%的AIH患者常规自身抗体检测呈阴性,该类患者血清IgG水平升高幅度常较小甚至正常,这给AIH的诊断带来很大困难,但肝组织学仍可见界面性肝炎、淋巴-浆细胞浸润、玫瑰花环等AIH特征性改变。因此,疑似自身抗体阴性AIH时,强烈建议行肝活组织检查以明确诊断,有时肝组织学表现是其唯一确诊依据。这类患者可予糖皮质激素单药治疗或联合治疗,对免疫抑制剂治疗应答常与典型AIH相似[42]。
约1/3的AIH患者在诊断时已存在肝硬化表现。活动性肝硬化患者仍有免疫抑制剂治疗的指征[17]。治疗方案以选择糖皮质激素单药治疗为宜,适当减少泼尼松(龙)初始剂量(20~30 mg/d),同时注意消化道出血和(或)感染等并发症的发生。AIH相关肝硬化患者应每6个月随访一次血清甲胎蛋白和腹部超声检查,以排除肝细胞癌的可能。当AIH相关肝硬化出现腹水等并发症,提示进入失代偿期。此阶段需仔细评估糖皮质激素可能的不良反应,如消化道出血、肺部感染和自发性细菌性腹膜炎的可能性。如疾病仍有明显活动,如血清转氨酶和TBil水平升高、血清IgG水平显著增高,在预防并发症的基础上可谨慎使用小剂量糖皮质激素(15~20 mg/d)口服,疾病好转后应快速减量至维持量(5~7.5 mg/d)。部分患者可获得生物化学应答,腹水等并发症好转而转入代偿期并获得长期缓解。如疗效不佳或无法耐受糖皮质激素治疗者,需尽早与肝移植中心联系进行肝移植治疗。
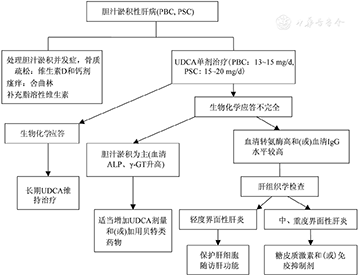
患者同时或在病程的不同阶段存在两种自身免疫性肝病的临床、血清学、组织学特征,称为自身免疫性肝病重叠综合征(简称重叠综合征),以AIH-PBC重叠综合征最为多见。IAIHG提出,AIH的积分系统最初目标是用于诊断AIH,并不适用于重叠综合征的诊断,主张将自身免疫性肝病分类为AIH、PBC、PSC、小胆管PSC,而重叠综合征并非独立疾病,目前缺乏明确的诊断标准和治疗方案[43]。重叠综合征诊断和处理思路可参见图4。
注: PBC为原发性胆汁性胆管炎;PSC为原发性硬化性胆管炎;UDCA为熊去氧胆酸;ALP为碱性磷酸酶;γ-GT为γ-谷氨酰转肽酶
AIH-PBC重叠综合征:PBC是一种以小叶间胆管非化脓性破坏性胆管炎为特征的自身免疫性肝病。由于疾病早、中期并无肝硬化表现,建议将疾病名称改为原发性胆汁性胆管炎(primary biliary cholangitis),仍保留PBC缩写,以更精确反映疾病特点,缓解患者心理压力[44]。AIH-PBC重叠综合征占所有PBC患者的5%~15%。2008年Chazouillères等[45]提出AIH-PBC重叠综合征诊断标准(巴黎标准),即AIH和PBC三项诊断标准中的各二项同时或者相继出现。AIH诊断标准包括:①血清ALT>5×ULN;②血清IgG≥2×ULN或血清ASMA阳性;③肝脏组织学提示中、重度界面性肝炎。PBC诊断标准包括:①血清ALP≥2×ULN或血清γ-GT≥5×ULN;②血清抗线粒体抗体(antimitochondrial antibodies, AMA)阳性;③肝脏组织学表现为非化脓性破坏性胆管炎。来自欧美的研究结果表明,巴黎标准用于诊断AIH-PBC重叠综合征的灵敏度和特异度分别达到92%和97%[46]。我国的研究结果表明,血清IgG水平≥1.3×ULN诊断激素敏感性PBC的灵敏度为60%,特异度为97%。88%激素敏感的PBC患者满足AIH简化积分系统的"确诊"标准(≥7分)[47]。
目前多数学者建议以泼尼松(龙)和UDCA进行联合治疗,可能有利于缓解病情,改善患者预后。泼尼松(龙)联合UDCA治疗不能缓解或泼尼松(龙)不良反应明显者,可加用免疫抑制剂如硫唑嘌呤。欧洲多中心研究结果显示,88例PBC-AIH重叠综合征患者中,37%对UDCA单剂治疗无应答,重度界面性肝炎是无应答的独立危险因素。UDCA和免疫抑制剂联合治疗在73%初始治疗或对UDCA单剂治疗无应答的患者中有效,而进展期纤维化与对联合治疗无应答有关。二线免疫抑制剂(环孢素A、他克莫司和MMF)可诱导54%的对初始免疫抑制剂无应答的患者获得生物化学缓解[48]。我国最新研究结果显示,糖皮质激素和UDCA联合治疗可显著改善重叠综合征患者的短期预后[49]。相关内容详见PBC的诊断和治疗共识。
AIH-PSC重叠综合征:PSC是一种较为少见的慢性胆汁淤积性肝病,其特征为肝内、外胆管弥漫性炎性反应和纤维化,引起胆管变形和节段性狭窄,病情呈进行性发展,最终导致胆汁性肝硬化及肝功能衰竭[50]。AIH-PSC重叠综合征的诊断标准是相加性的,即在明确的PSC诊断基础上,同时存在AIH特征性临床表现(血清转氨酶和IgG水平显著升高)和肝组织学特征(中、重度界面性肝炎等)。AIH-PSC重叠综合征患者UDCA(15~20 mg/kg)联合糖皮质激素[泼尼松(龙)]治疗,可改善患者血清生物化学指标,但组织学及长期疗效未得到证实。相关内容详见PSC的诊断和治疗共识。
AIH合并慢性病毒性肝炎常难以识别和确诊,容易造成延误诊断,大多数患者诊断时合并肝硬化。在25例合并病毒性肝炎的AIH患者中,20例(80%)合并HCV、5例(20%)合并HBV感染[51]。我国AIH患者HBsAg阳性率仅为0.83%,PBC患者为1.02%,显著低于非自身免疫性疾病患者(4.58%)[52],但不能完全排除在HBsAg阳性患者中自身免疫性肝病被低估的可能性。肝组织学对于诊断和鉴别诊断显得非常重要,综合诊断积分系统显然优于简化标准[2]。
AIH合并HBV感染者先以核苷(酸)类似物口服抗病毒治疗,然后再开始免疫抑制剂治疗。而AIH合并HCV感染患者首先考虑免疫抑制剂进行治疗,获得生物化学缓解后再考虑使用长效α干扰素等抗病毒治疗。直接抗病毒药物治疗方案的出现为HCV合并AIH患者的处理带来新的机遇,小分子抗病毒药物和糖皮质激素可同时使用。
AIH患者妊娠过程中,可予小剂量泼尼松(龙)5~10 mg/d维持治疗。AIH患者有较高的胎儿流产及早产的可能性,胎儿死亡率达19%,大多发生在孕20周内,产妇死亡率约为3%。妊娠过程中母体的免疫抑制可保护嵌合胎儿,在分娩后AIH可复燃或出现加重趋势。因此,应在分娩后加大糖皮质激素的用量,以防止复发或反跳。最近的一项研究结果显示,在53名妇女的81次妊娠中,41%的妊娠发生在肝硬化条件下,75%的患者在维持治疗中。活产率为73%,早产率为20%,11%的婴儿需进入特护病房。在妊娠前AIH控制较差或妊娠期不治疗与AIH复燃有关,而AIH复发与孕妇出现失代偿、婴儿进入特护病房的危险性有关[53]。目前没有关于因使用硫唑嘌呤治疗AIH而引起胎儿畸形的报道,但已证实硫唑嘌呤对小鼠有致畸作用,所以建议AIH患者在怀孕期间停用硫唑嘌呤。美国食品药品管理局将硫唑嘌呤定为妊娠D级,故建议尽量在妊娠期间停用。
20%的成人AIH在60岁以后发病,发病常更为隐匿,易被漏诊[54]。一项Meta分析共纳入10项回顾性研究,对264例老年AIH患者(≥60岁)和592例年轻患者进行系统分析。老年患者中无症状者、诊断时已存在肝硬化、HLA-DR4呈阳性的概率显著高于年轻患者。糖皮质激素可用于老年患者,应答相对较好且停药后复发概率较低[55]。布地奈德可以考虑用于这一特殊人群中。在老年AIH患者中,预防骨质疏松尤为重要,应鼓励常规锻炼,服用钙剂(1~1.5 g/d)和维生素D3 (400 IU/d)。在已经有骨质疏松的患者可考虑使用二磷酸盐制剂。应进行基线骨密度测定,并每年复查以观察严重程度和疗效[56]。
1型AIH常在青春期前、后发病,而2型AIH发病较早,甚至可在婴儿期发病。15%的1型AIH和25%的2型AIH可表现为血清IgG水平正常。1型AIH易出现肝硬化表现,而2型AIH更易表现为急性肝功能衰竭。两种类型中SLA阳性的患儿疾病严重程度较高,并易复发。20%患儿合并其他自身免疫性疾病包括甲状腺炎、白癜风、1型糖尿病、肾病综合征等。IAIHG开发的综合和简化积分系统已广泛用于成人AIH的诊断,但并未考虑到儿童患者的特殊性,在儿童患者中自身抗体的效价往往低于成人患者。而且这些积分系统也不能区分AIH和儿童中较常见的自身免疫性硬化性胆管炎[57]。
儿童AIH的治疗包括泼尼松龙1~2 mg/(kg·d)(最大不超过40 mg/d),随着转氨酶水平下降,在4~8周内减量至维持剂量(根据患儿的体质量和年龄2.5~5 mg/d维持)[17]。大多数患儿在最初2个月内血清转氨酶的降幅达80%以上,但获得完全生物化学缓解可能需数月。在治疗的最初6~8周,应经常检测肝功能以便每周进行糖皮质激素的剂量调整。英国国王学院医院一般在转氨酶停止下降或出现明显糖皮质激素不良反应时加用硫唑嘌呤,以0.5 mg/(kg·d)剂量开始,在无明显不良反应情况下逐渐加量至最大量[2 mg/(kg·d)][58]。应注意硫唑嘌呤的肝毒性,特别是在胆汁淤积患儿中。最佳疗程尚未确定,仅在肝组织炎性反应缓解后才能成功停药。因此,肝功能生物化学指标和IgG水平正常1~2年、自身抗体阴性或效价低时可行肝组织学检查,如显示汇管区轻微炎性反应或无炎性反应时才能停药。20%的1型AIH患儿可成功停药,而2型AIH极少停药成功。在儿童期,监测IgG水平和自身抗体效价非常关键,两项中任何一项的波动预示疾病活动。对于IgG水平高的患儿,其下降能可靠、客观且廉价地监测疾病控制情况。对免疫抑制剂治疗应答良好的患儿预后较好,大多数可长期生存且生存质量较好。约8.5%的患儿尽管接受治疗,在8~14年后仍出现终末期肝病,需行肝移植术。
推荐意见23:急性起病的AIH(慢性疾病基础上的急性发作或无慢性疾病基础的急性AIH)应及时启动糖皮质激素治疗,以防止急性肝功能衰竭的发生(1C)。
推荐意见24: AIH相关急性肝功能衰竭可先予短期静脉滴注甲泼尼松龙(一般剂量为40~60 mg/d)治疗,若患者1周内病情无明显改善甚至有恶化者需考虑肝移植术(1C)。
推荐意见25: AIH伴胆汁淤积表现者需排除PBC和PSC等胆管疾病,在泼尼松(龙)治疗的基础上可联合使用UDCA[13~15 mg/(kg·d)](1C)。
推荐意见26:对于自身抗体阴性而肝组织学检查呈典型AIH表现者,在严格排除其他病因后可考虑进行糖皮质激素的试验性治疗,如应答良好支持AIH诊断(1C)。
推荐意见27: AIH特别是合并肝硬化的患者应每6个月检测1次血清甲胎蛋白和腹部超声检查,以筛查肝细胞癌(1C)。
推荐意见28:活动性AIH相关肝硬化失代偿期患者,在预防并发症的基础上可谨慎使用小剂量UDCA(一般剂量为15~20 mg/d)口服,疾病好转后应快速减量至维持量(一般剂量为5~7.5 mg/d)(1C)。
推荐意见29:具有PBC或PSC显著特点的AIH患者需考虑重叠综合征诊断,并予UDCA和免疫抑制剂的联合治疗(1C)。
推荐意见30: AIH合并HBV感染者先以核苷(酸)类似物口服抗病毒治疗,然后再开始免疫抑制剂治疗。AIH合并HCV感染者有条件者先予直接抗病毒药物治疗,再进行免疫抑制剂治疗。在AIH未控制之前慎用α干扰素抗病毒治疗(1C)。
推荐意见31:在AIH患者妊娠过程中,可予小剂量泼尼松(龙)(一般剂量为5~10 mg/d)维持治疗。在患者分娩前2周或分娩后应适当加大糖皮质激素以降低复发风险(1C)。
推荐意见32:老年AIH患者发病隐匿,一般对糖皮质激素应答较好,复发率低,但在治疗过程中需及时发现和预防骨质疏松症(1C)。
推荐意见33:儿童AIH患者确诊后即应启动免疫抑制剂治疗,推荐泼尼松(龙)和硫唑嘌呤联合治疗方案或泼尼松(龙)单药治疗方案(1C)。
AIH患者在获得生物化学缓解后一般预后较好、生存期接近正常人群。预后不佳的危险因素主要包括诊断时已有肝硬化和治疗后未能获得生物化学缓解。我国的研究结果显示,合并其他系统自身免疫性疾病、肝内胆管损伤和诊断时MELD评分较高者与治疗应答和预后不佳有关[59]。日本AIH患者的10年生存率为94.9%,肝病相关病死率为3.4%。经历2次以上复发的患者较获得持续缓解者生存期缩短[60]。新西兰患者10年生存期在不同年龄段分别为80%~100%,在6个月内未能获得ALT复常、血清白蛋白<36 g/L、入选时年龄≤20岁或≥60岁是肝病相关死亡的危险因素[61]。英国患者的10年生存率为84%,而20年生存率仅为48%。肝组织学证实肝硬化、入选时失代偿、在治疗后1年未能使ALT复常以及每10年复发次数多于4次是预后不良的危险因素[62]。丹麦全国的AIH患者(1 721例)的10年生存率为73.6%,肝脏相关病死率为10.2%。男性和入选时肝硬化与病死率增高和肝细胞癌发生有关。在有肝组织学资料的患者(1 318例)中,28.3%的患者存在肝硬化,肝细胞癌的10年累积发生率为0.7%。在诊断后的1年内,AIH患者病死率为普通人群的6倍,之后病死率仍为2倍。10年累积病死率为26.4%,其中38.6%的死亡与肝病相关,包括3.6%死于肝细胞癌[4]。德国的研究结果表明,年龄<18岁、初诊时已有肝硬化、SLA阳性是短期和长期预后不佳的主要危险因素[18]。总之,诊断时的肝硬化和治疗应答是决定患者长期预后的两个最重要危险因素。
尽管近年来在AIH的诊断、治疗和发病机制方面取得了长足的进步,关于AIH仍面临诸多问题和挑战[41]。虽然2型AIH的自身抗原(细胞色素P450 2D6)已被鉴定,1型AIH的自身抗原仍未确定,大大阻碍了自身抗原特异性治疗的开发。目前尚缺乏与人类AIH相似的动物模型,发病机制研究进展较缓慢。AIH的诊断较为复杂,是排除诊断基础上的综合诊断,尚缺乏特异性诊断标志物和诊断时预测高危患者的标志物。目前,AIH的治疗仍为全身免疫抑制剂的应用,优化治疗方案或二线药物的选择有待临床验证。抗原特异性的免疫调控细胞如调节性T淋巴细胞和髓系免疫抑制细胞回输,可能是具有前景的AIH治疗手段之一[63,64]。最后,由于AIH患者数有限,开展我国多中心的临床合作研究显得尤为重要。
参加AIH诊断和治疗共识撰写的专家人员名单(排名不分先后,按姓氏汉语拼音为序)
陈成伟、陈晓宇、成军、窦晓光、段钟平、范建高、傅青春、高春芳、韩英、侯金林、胡和平、胡锡琪、贾继东、刘玉兰、陆伦根、马雄、茅益民、苗琪、南月敏、邱德凯、任红、王贵强、王吉耀、王建设、王绮夏、王宇明、魏来、谢青、谢渭芬、许建明、闫惠平、杨长青、尤红、曾民德、张文宏、周新民、庄辉、诸葛宇征、邹晓平
























