
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Klinefelter综合征又称先天性生精小管发育不全,由染色体核型异常引起。该病最常见的染色体核型异常为47,XXY,约占全部患者的80%;其他的20%则表现一定的差异,主要是有附加的X或Y,或镶嵌型,如48,XXXY;49,XXXXY;48,XXYY;49,XXXYY;46,XY/47,XXY;46,XY/45,X/47,XXY;46,XX/47,XXY;46,XY/46,XX/47,XXY[1],目前对于少见类型的Klinefelter综合征报道较少。由于少见类型Klinefelter综合征发病不典型,因此诊断过程大多曲折,笔者收治1例48,XXYY型Klinefelter综合征,现报告如下。
患者男性,45岁,因"身材高大及无第二性征发育30余年"于2014年7月6日就诊。患者青春期比同龄人高大,无第二性征发育及性功能,但一直未重视及就医。2012年开始出现头昏、头痛、心悸。入院体格检查:身高188 cm,五官正常,女性表征,皮肤细腻,双颧隆突,唇舌肥厚,下颌突出,牙齿稀疏。甲状腺无肿大。外生殖器为幼稚型,阴毛腋毛稀少;阴茎长度3.2 cm,睾丸约鹌鹑蛋大小,阴囊皮肤松薄,色红。辅助检查:血常规、肝肾功、心房利钠肽、肌钙蛋白未见明显异常;促肾上腺皮质激素44.05 pg/ml(参考值范围5~60 pg/ml,下同);甲状腺功能:FT3 3.97 pmol/L(3.1~6.8)、FT4 14.95 pmol/L(12~22)、TSH 2.26 μIU/ml(0.27~4.2);性激素六项:E2 22.0 pg/ml(20~75)、孕酮0.47 ng/ml(0.1~0.84)、FSH 57.53 IU/L(1.27~19.26),LH 26.0 IU/L(1.24~8.62)、催乳素15.15 ng/ml(2.64~13.13)、睾酮0.48 ng/ml(1.75~7.81);口服葡萄糖糖耐量(OGTT)及生长激素抑制试验未见异常,胰岛素及C肽高峰后延至2 h(表1)。患者性激素水平提示睾酮水平低下,但FSH及LH均升高,表明患者睾丸功能不全;同时行促性腺激素释放激素(GnRH)兴奋试验提示经醋酸曲普瑞林100 μg刺激后LH升高5倍左右,表明垂体功能正常(表2)。人绒毛膜促性腺激素(HCG)兴奋试验表明基础睾酮水平低,同时经HCG刺激后无反应,呈现低平曲线(表3)。以上实验表明,患者睾丸功能不全,垂体功能正常,雄激素低下的原因在睾丸。
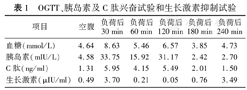
OGTT、胰岛素及C肽兴奋试验和生长激素抑制试验
OGTT、胰岛素及C肽兴奋试验和生长激素抑制试验
| 项目 | 空腹 | 负荷后30 min | 负荷后60 min | 负荷后120 min | 负荷后180 min | 负荷后240 min |
|---|---|---|---|---|---|---|
| 血糖(mmol/L) | 4.64 | 8.63 | 5.46 | 6.57 | 3.85 | 4.73 |
| 胰岛素(mIU/L) | 4.58 | 33.75 | 15.92 | 31.17 | 2.42 | 2.70 |
| C肽(ng/ml) | 1.31 | 5.95 | 4.15 | 5.49 | 2.01 | 1.50 |
| 生长激素(μIU/ml) | 0.49 | 3.70 | 0.21 | 0.05 | 0.76 | 3.49 |
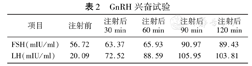
GnRH兴奋试验
GnRH兴奋试验
| 项目 | 注射前 | 注射后30 min | 注射后60 min | 注射后90 min | 注射后120 min |
|---|---|---|---|---|---|
| FSH(mIU/ml) | 56.72 | 63.37 | 65.93 | 90.97 | 89.43 |
| LH(mIU/ml) | 20.09 | 72.52 | 88.59 | 105.95 | 103.81 |
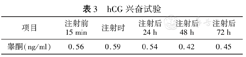
hCG兴奋试验
hCG兴奋试验
| 项目 | 注射前15 min | 注射时 | 注射后24 h | 注射后48 h | 注射后72 h |
|---|---|---|---|---|---|
| 睾酮(ng/ml) | 0.56 | 0.59 | 0.54 | 0.42 | 0.45 |
心脏超声:左房稍增大,二尖瓣、三尖瓣、肺动脉瓣轻度反流;左室舒张早期驰张功能降低,左室假腱索。睾丸超声:睾丸容积约6 ml。蝶鞍MRI:鞍区和垂体平扫未见明确异常;左侧基底节和额顶叶皮层下异常信号,缺血灶?头颈MRI:双侧脑室旁多发性腔隙灶,缺血灶?心电图:窦性心动过缓,I度房室传导阻滞。动态心电图:窦性心动过缓,短阵房性心动过速、室上性心动过速。染色体:48,XXYY(图1)。诊断为48,XXYY Klinefelter综合征,由于患者有心血管症状及表现,予以阿司匹林对症治疗,同时予以安特尔,40 mg bid,服药半年后患者病情平稳,无特殊不适。
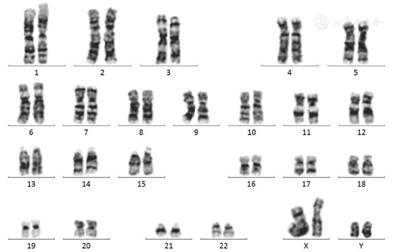
1942年Klinefelter首次报道此病,其中48,XXYY型Klinefelter综合征是一种非常罕见的遗传疾病,其发病率为1/50 000[2]。Klinefelter综合征主要表现为男性第二性征发育差,睾丸发育不全或原发小睾丸症等,并且大部分无法生育,有女性化表现,如无胡须、体毛少、阴毛分布如女性,阴茎龟头小等,约25%的患者有乳房发育,也叫类宦官综合征。而48,XXYY Klinefelter综合征主要表现为身材高大、智力障碍、精神障碍和骨骼血管的病变[3]。本例患者存在身材高大及四肢长,这种临床表现极易误导往生长激素过多的方向去考虑疾病。Klinefelter综合征是常见的先天性性腺发育异常,其主要病因为睾丸曲细精管发育不良,该病需染色体检查确诊,我们对该患者行染色体检查,结果显示染色体为48,XXYY,故明确诊断为48,XXYY Klinefelter综合征。
有文献报道48,XXYY作为特殊类型的Klinefelter综合征,除了符合Klinefelter综合征的常见临床表现外,还表现为行为及精神障碍[4],颅脑MRI可见海马区的病变。"海马区"是大脑皮质的一个内褶区,在侧脑室底部绕脉络膜裂形成一弓形隆起,它由两个扇形部分组成,有时将两者合称海马结构。本例患者蝶鞍MRI及颅脑MRI均提示侧脑室旁的缺血灶改变可能,虽然患者行为异常表现不明显,但存在的头昏、头痛等表现可能与磁共振所见的侧脑室缺血灶改变有关。许多前瞻性研究表明,低水平的睾丸激素可以预测2型糖尿病的发生[5],Grossmann等[6]的研究提示睾酮和胰岛素抵抗之间存在反比关系。有报道发现Klinefelter综合征患者发生糖尿病是由于遗传因素、自身免疫机制或内分泌异常,其中胰岛素抵抗的作用较大[7,8]。本例患者胰岛素释放提示2 h水平仍然较高。部分文献报道[9],48,XXYY Klinefelter综合征患者FSH水平一般比LH水平高。本例患者GnRH兴奋试验提示FSH水平低于LH水平,提示睾丸功能障碍来自于睾丸支持细胞,该临床差异是否为47,XXY与48,XXYY的鉴别点,目前暂无临床报道,尚待更多的临床观察及研究。部分患者存在牙齿稀疏[8]。本例患者在入院查体即发现了该临床特征,因此临床工作中详细的体格检查显得非常必要。先天性心脏疾病也常见于48,XXYY Klinefelter综合征[10],其发病率约为19.4%[11],本例患者相关辅助检查也提示存在心脏损害,但缺乏患者早年的体检资料,因此不能确定其心脏损害是本病导致还是其他原因所致。目前由于大多数医院无法开展染色体检查而令许多患者漏诊或误诊,因此常规的性激素水平测定以及HCG、GnRH兴奋试验就显得尤为重要,对于身材高大、四肢长,同时性腺发育不全者应考虑到该病的可能,可通过上述检查帮助诊断,同时颅脑MRI、胰岛素及C肽释放试验等检查也可作为支持依据。
Klinefelter综合征以雄激素(十一酸睾酮或安特尔)终身替代治疗为主,小剂量起始,根据雄激素水平逐渐滴定至维持剂量,而不以FSH和LH降低到正常水平作为雄激素剂量和疗效判断的指标。大多数患者通过单纯的雄激素替代治疗不能正常生育,但Palermo等[12]报道应用睾丸精子提取联合单精子卵浆内注射辅助生殖技术,可以生育。早期发现该病并给予雄激素替代治疗可维持患者正常的第二性征及性功能,采取一些新技术治疗甚至可让部分患者获得生育能力。青春期密切关注第二性征发育以及婚前检查有利于本病早期发现。

























