
分析CFHR1、CFHR3基因异常的非典型溶血尿毒综合征(aHUS)患儿的临床特征。
收集13例aHUS患儿的临床、病理资料及CFHR1、CFHR3基因检测结果,回顾性分析其临床特点、病理特点及治疗和预后,结合相关基因的检测结果进行临床特征分析。
13例aHUS患儿纳入研究。其中男9例,女4例;年龄1.5~13.0岁。13例患儿除具有特征性的HUS三联征外,均有恶心、呕吐、腹痛或腹胀等胃肠道症状,但均无腹泻;均表现为大量蛋白尿,补体C3均下降;12例合并高血压。3例患儿进行了肾脏穿刺活检。患儿在确诊后均采用了血浆置换治疗,治疗后1例患儿为慢性肾脏病3期,余肾功能均恢复正常,蛋白尿消失或明显减轻,补体C3均恢复正常。在13例患儿中,CFH基因均未发现突变,6例有CFHR1/CFHR3基因缺失,1例CFHR1基因纯合缺失,2例CFHR1基因纯合突变,1例CFHR1基因杂合突变,2例CFHR1杂合缺失,1例CFHR1、CFHR3基因拷贝数增加。
CFHR1、CFHR3基因异常的aHUS患儿肾功能损害重,补体下降突出,应以强化血浆置换为主要治疗方式,需要尽早进行,且血浆置换次数多,血浆用量大,且相对易复发。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
溶血尿毒综合征(hemolytic uremic syndrome,HUS)是一种以微血管性溶血性贫血、血小板减少和急性肾衰竭为主要临床特征的疾病。临床上根据HUS发病诱因分为典型HUS和非典型HUS(atypical HUS,aHUS)。典型HUS又称腹泻相关性HUS,而aHUS则指非腹泻相关HUS,是一种非常罕见的、危及生命的进展性疾病,常与遗传因素有关。aHUS近年来有增多趋势,其预后差,进展至终末期肾病比例高。aHUS被认为是多因素疾病,由基因(补体调控蛋白基因突变等)易感性与感染或细胞毒素等环境因素共同激活补体系统,引起aHUS发病。约50%的aHUS患者由于补体系统的基因缺陷引起[1]。H因子参与aHUS的发病主要表现为以下2种方式:(1)H因子基因纯合或杂合缺陷造成血浆H因子水平下降和/或功能缺陷;(2)由于抗H因子自身抗体的存在,抑制了H因子与C3b、CRP及内皮细胞结合的能力,使得补体旁路途径过度活化而引起组织损伤[2]。本研究选取了13例H因子相关蛋白(CFHR)1、CFHR3基因异常的aHUS,对其临床特点、病理特点及治疗和预后进行分析和总结,旨在分析CFHR1、CFHR3基因异常的aHUS患儿的临床特征。
收集2012年1月至2015年12月在首都医科大学附属北京儿童医院肾病科住院的aHUS患儿资料,同时符合以下3条标准:(1)年龄<18岁;(2)临床符合aHUS诊断标准[3];(3)补体CFHR1、CFHR3基因异常。排除标准:CFHR1、CFHR3基因正常的aHUS。所有临床资料及标本的获取均已获得患儿家长知情同意,签署知情同意书。本研究已通过医院医学伦理委员会批准。
采集患儿的临床资料,主要包括一般情况(性别和诊断年龄等),临床表现,肾功能,24 h尿蛋白定量,补体C3、C4等实验室检查,其中3例患儿进行了肾组织活检,治疗及预后随访资料。患儿的基因检测:检测的基因包括CFH、CFHR1和CFHR3。
(1)测序:①标本采集:在知情同意的前提下,空腹采集患者乙二胺四乙酸(EDTA)抗凝血4 mL进行基因分析。②基因扩增:取患者新鲜全血4 mL,使用BloodGen Midi Kit(CWBIO,China)提取患者全基因组DNA,操作按照试剂盒说明书进行。根据各基因序列设计引物,采用PCR方法进行扩增。PCR反应条件为:95 ℃预变性5 min;95 ℃变性30 s,50~65 ℃退火30 s,72 ℃链延伸1 min,扩增30个循环;最后72 ℃补充延伸10 min。PCR的体系均为50 μL。
基因序列分析:基因各外显子的PCR扩增产物,用ABI 3730XL测序仪测序,测序引物采用原PCR引物并进行测通。
基因序列分析采用DNASTAR软件进行序列分析和比对。比对的mRNA模板:CFH(NM_000186.3),CFHR1(NM_002113.2),CFHR3(NM_021023.5)。
(2)CFHR1和CFHR3基因外显子拷贝数测定:分别在CFHR1基因第2、3、5外显子,CFHR3基因第1、2、6外显子设计特异的荧光定量引物,以人血清清蛋白基因(HAS)为内参基因,采用荧光定量PCR方法进行扩增,以ALB基因为参照基因。PCR反应条件:50 ℃ 2 min;95 ℃ 2 min;95 ℃ 15 s,60 ℃ 40 s,扩增40个循环;PCR的体系均为10 μL。
对正常对照样本(同期首都医科大学附属北京儿童医院保健科健康患儿体检剩余标本)和先证者做荧光定量PCR,每个反应管做2次重复,同时做每对引物的标准曲线,以获取其扩增效率。分析数据,得到目标基因与内参基因拷贝数的比值。
2012年1月至2015年12月共收治13例符合入选标准aHUS患儿,均符合补体CFHR1、CFHR3基因异常,全部纳入本研究。其中男9例、女4例,年龄1.5~13.0岁。患儿均无肾脏疾病及血栓性微血管病家族史。其中3例患儿进行了肾组织活检,并进行了肾活检组织免疫荧光染色和电镜检查。
本组患儿除表现为微血管病性溶血性贫血、血小板减少和急性肾衰竭等特征性的HUS表现外,均有恶心、呕吐、腹痛或腹胀等胃肠道症状,但无腹泻。13例患儿均伴高血压(超过同年龄、性别组 ±2 s及以上)。
±2 s及以上)。
本组患儿均有肾功能下降、贫血和网织红细胞增高、血小板减少,外周血可见红细胞碎片,抗人球蛋白试验阴性。患儿均表现为大量蛋白尿,补体C3均下降,补体C4均正常。临床资料、实验室检查治疗情况,见表1。
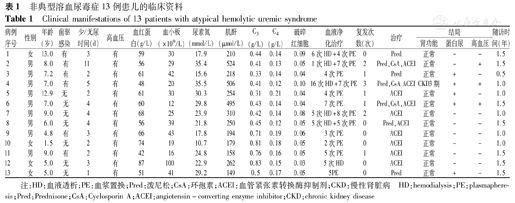
非典型溶血尿毒症13例患儿的临床资料
Clinical manifestations of 13 patients with atypical hemolytic uremic syndrome
非典型溶血尿毒症13例患儿的临床资料
Clinical manifestations of 13 patients with atypical hemolytic uremic syndrome
| 病例序号 | 性别 | 年龄(岁) | 前驱感染 | 少/无尿时间(d) | 高血压 | 血红蛋白(g/L) | 血小板(×109/L) | 尿素氮(mmol/L) | 肌酐(μmol/L) | C3(g/L) | C4(g/L) | 破碎红细胞 | 血液净化治疗 | 复发次数(次) | 治疗 | 结局 | 随访时间(年) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 肾功能 | 蛋白尿 | 高血压 | |||||||||||||||||
| 1 | 女 | 13.0 | 有 | 3 | 有 | 59 | 30 | 17.9 | 210 | 0.44 | 0.14 | 0.09 | 6次HD+4次PE | 0 | Pred | 正常 | - | - | 1.5 |
| 2 | 男 | 8.0 | 有 | 11 | 有 | 56 | 29 | 35.4 | 524 | 0.41 | 0.13 | 0.05 | 1次HD+7次PE | 2 | Pred,CsA,ACEI | 正常 | - | + | 1.5 |
| 3 | 男 | 7.2 | 有 | 2 | 有 | 61 | 42 | 15.6 | 218 | 0.33 | 0.14 | 0.04 | 4次PE | 1 | Pred | 正常 | + | - | 0.5 |
| 4 | 男 | 7.0 | 有 | 5 | 有 | 48 | 20 | 35.5 | 506 | 0.41 | 0.12 | 0.10 | 16次HD+7次PE | 3 | Pred,CsA ACEI | CKD3期 | + | + | 1.0 |
| 5 | 男 | 12.9 | 无 | 2 | 有 | 61 | 30 | 30.3 | 254 | 0.31 | 0.21 | 0.04 | 4次PE | 1 | ACEI | 正常 | + | - | 1.0 |
| 6 | 男 | 7.0 | 无 | 4 | 有 | 60 | 12 | 29.8 | 495 | 0.43 | 0.14 | 0.04 | 7次PE | 1 | Pred,CsA,ACEI | 正常 | + | + | 1.5 |
| 7 | 男 | 9.0 | 无 | 4 | 有 | 68 | 25 | 23.9 | 310 | 0.42 | 0.14 | 0.08 | 5次HD+8次PE | 2 | ACEI | 正常 | - | - | 1.0 |
| 8 | 男 | 6.0 | 无 | 4 | 有 | 56 | 39 | 21.8 | 250 | 0.45 | 0.12 | 0.05 | 5次HD+5次PE | 0 | Pred,ACEI | 正常 | - | - | 1.5 |
| 9 | 男 | 4.8 | 有 | 3 | 有 | 66 | 43 | 17.8 | 194 | 0.71 | 0.19 | 0.06 | 3次PE | 0 | ACEI | 正常 | - | - | 1.0 |
| 10 | 女 | 1.5 | 无 | 2 | 有 | 74 | 19 | 10.7 | 179 | 0.81 | 0.18 | 0.05 | 2次PE | 0 | ACEI | 正常 | - | - | 1.0 |
| 11 | 男 | 9.0 | 有 | 2 | 有 | 42 | 16 | 24.8 | 158 | 0.76 | 0.16 | 0.05 | 5次PE | 1 | ACEI | 正常 | - | - | 1.5 |
| 12 | 女 | 5.0 | 无 | 3 | 有 | 87 | 100 | 22.9 | 262 | 0.83 | 0.15 | 0.03 | 5次HD | 0 | ACEI | 正常 | - | - | 1.5 |
| 13 | 女 | 5.0 | 无 | 1 | 有 | 51 | 41 | 29.2 | 149 | 0.5 | 0.17 | 0.05 | 5PE | 0 | Pred | 正常 | + | - | 1.5 |
注:HD:血液透析;PE:血浆置换;Pred:泼尼松;CsA:环孢素;ACEI:血管紧张素转换酶抑制剂;CKD:慢性肾脏病 HD:hemodialysis;PE:plasmapheresis;Pred:Prednisone;CsA:Cyclosporin A;ACEI:angiotensin-converting enzyme inhibitor;CKD:chronic kidney disease
3例患儿(例2、例6、例7)进行了肾脏穿刺活检,光镜检查均可见肾小球系膜细胞和基质不同程度弥散或节段增生,毛细血管襻内皮细胞增生及肿胀;小动脉管壁增厚,部分小动脉壁葱皮样增生,可见毛细血管腔内淤血伴微血栓形成。2例患儿免疫荧光染色可见系膜区轻度IgG、IgM和C3沉积,1例免疫荧光染色均为阴性。电镜检查可见毛细血管内皮细胞肿胀,基底膜内疏松层增厚,见图1。
患儿在确诊后除1例(例12)外均采用了血浆置换治疗,1例患儿为慢性肾脏病(CKD)3期,余肾功能均恢复正常,蛋白尿消失或明显减轻,补体C3恢复正常。
患儿基因检测结果,见表2。在13例患儿中,CFH基因均未发现突变,检出CFHR1和CFHR3基因的纯合缺失6例,包括CFHR1基因的缺失、突变和2个基因的缺失,CFHR1基因的杂合缺失,CFHR1基因的杂合突变和CFHR1、CFHR3基因拷贝数增加。
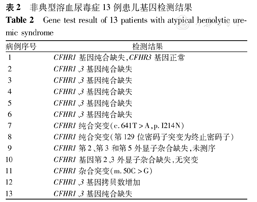
非典型溶血尿毒症13例患儿基因检测结果
Gene test result of 13 patients with atypical hemolytic uremic syndrome
非典型溶血尿毒症13例患儿基因检测结果
Gene test result of 13 patients with atypical hemolytic uremic syndrome
| 病例序号 | 检测结果 |
|---|---|
| 1 | CFHR1基因纯合缺失,CFHR3基因正常 |
| 2 | CFHR1、3基因纯合缺失 |
| 3 | CFHR1、3基因纯合缺失 |
| 4 | CFHR1、3基因纯合缺失 |
| 5 | CFHR1、3基因纯合缺失 |
| 6 | CFHR1、3基因纯合缺失 |
| 7 | CFHR1纯合突变(c.641T>A,p.I214N) |
| 8 | CFHR1纯合突变(第129位密码子突变为终止密码子) |
| 9 | CFHR1第2、第3和第5外显子杂合缺失,未测序 |
| 10 | CFHR1基因第2、3外显子杂合缺失,无突变 |
| 11 | CFHR1杂合突变(m.50C>G) |
| 12 | CFHR1、3基因拷贝数增加 |
| 13 | CFHR1、3基因纯合缺失 |
补体系统是一个由30余种蛋白组成的先天性免疫系统。补体激活过程依据其起始顺序不同,分为经典途径、凝集素途径和旁路途径。正常情况下,补体激活及其末端效应均处于严密调控之下,从而有效地维持机体的自稳功能。补体旁路途径可识别自己与非己,体内不断产生低水平血清补体片段C3b,少数C3b可以随机方式与颗粒表面形成共价键。若沉积在自身细胞,C3b可被补体调控蛋白迅速裂解灭活,中止补体旁路途径激活的级联反应。在这个过程中起到关键作用。反之,若缺失这些补体调控蛋白,C3b沉积使补体级联反应持续放大,从而使血管内皮细胞遭受损伤,进而导致aHUS[4]。
CFH基因位于1q32,全长94 kb,有23个外显子,CFH是一种主要由肝脏产生的糖蛋白,含有1 231个氨基酸,由20个补体调控蛋白重复单位(complement control protein repeats,CCPs)组成,CCPs又称为短同源重复序列(short consensus repeats,SCR)。CFH编码功能相关和结构相似的7种蛋白:CFH、CFHL-1、补体因子H相关蛋白CHR1、CHR2、CHR3、CHR4和CHR5。在aHUS患者中存在H因子水平的降低或缺如,目前认为其主要原因包括H因子基因纯合或杂合缺陷,纯合突变时血浆H因子常降至正常水平的10%以下,杂合缺陷患者的H因子水平为正常水平的50%左右,H因子基因突变主要发生在SCR的19~20区域,为单个氨基酸的突变,虽未改变H因子的空间结构,但可以降低H因子与相应配体以及内皮细胞的结合能力[5]。
对H因子突变导致aHUS的小鼠模型的研究发现了H因子的缺陷导致aHUS[6]。因为H因子与CFHR基因在染色体上位置临近,经常发生非等位基因同源重组,出现大的染色体片段丢失,也可能导致aHUS的发生[6,7,8,9]。目前已知的H因子相关缺陷包括H因子突变,非等位基因同源重组、CFHR异常和抗H因子抗体的形成等。本组aHUS发病呈散发,病情重,需多次血浆置换强化治疗才能缓解,且易复发。在aHUS患者中CFH基因突变的患者约占30%,基因内突变多为错义突变。在aHUS患者中最常见的基因异常主要有CFHR1缺失、CFHR1/CFHR3联合缺失、CFHR5突变等。26.5%的患者有CFHR3和CFHR1缺失[4]。本研究中的患者血检结果均为抗H因子抗体阳性,其中6例(46.2%)有CFHR1/CFHR3的基因缺失,3例有CFHR1基因的纯合缺失或突变,这些患者肾功能损害重,补体下降突出,血浆置换次数多,血浆用量大,且相对易复发。3例有CFHR1基因的杂合缺失或突变。1例发现CFHR1、CFHR3基因拷贝数增加。CFHR1缺失后可能会使H因子介导的作用增强,促进补体失活[10,11]。目前文献报道90%存在抗H因子抗体的患者合并存在CFHR1/CFHR3的联合缺失(与编码CFHR1和CFHR3的基因缺失有关),具体机制目前尚不明确[12]。
2005年,Dragon-Durey等[13]报道了在aHUS患者中存在抗H因子自身抗体及CFHR的缺失,主要见于青少年男性,发生率为6%~10%。抗H因子抗体结合在H因子的SCR19~20区域,从而抑制H因子与C3b以及细胞表面结合,影响细胞表面补体旁路途径的调控,但并未影响血液中H因子作为辅助因子的活性,其作用类似于H因子突变[2]。Skerka等[14,15]报道儿童HUS中25%的患者存在CFH基因突变和自身抗体,并阐述了一个导致非典型HUS的重要机制:即CFHR血浆蛋白缺乏和自身抗体阳性的HUS(DEAP-HUS),表现为血浆中CFHR缺失(通常是CFHR1和CFHR3)并存在抗H因子自身抗体。近期研究显示10%~15%的aHUS患者中存在抗H因子自身抗体,同时在部分患者中还存在CFHR1和CFHR3基因的缺失,形成DEAP-HUS[16,17]。研究发现体内存在CFH自身IgG抗体的一组患者中,有CFHR/CFHR3基因缺陷。Strobel等[18]提供了更充足的证据表明自身免疫性HUS中CFH自身抗体对细胞表面CHF的抑制作用。
2009年欧洲aHUS研究和初始治疗指南建议aHUS首发症状出现后24 h内进行,伴随支持治疗(输血、血液透析、降血压等)[3]。aHUS中的一些病例研究提示激素联合免疫抑制剂治疗似乎对此类HUS有效。本研究表明及时对患者进行基因分析、血清H因子水平及H因子抗体检测,除了解疾病的致病机制外,还为疾病的治疗提供了指导,从而改善患儿的预后。

























