
探讨p16基因缺失在成人Ph染色体阳性急性淋巴细胞白血病(Ph+ ALL)中的临床意义。
回顾性分析80例Ph+ALL伴p16基因缺失患者的临床特征、免疫表型、细胞遗传学、分子生物学改变及其预后。
31.3% Ph+ALL患者合并p16基因缺失;p16基因缺失组与非缺失组相比,初诊时高白细胞计数(WBC≥30×109/L)更常见,高表达CD20,更易出现附加染色体异常,其中以累及7、8、19号染色体以及der(22)较为常见;两组诱导缓解率比较差异无统计学意义(P=0.033),p16基因缺失组患者治疗3个疗程后获BCR-ABL融合基因主要分子学反应(MMR)率和完全分子学反应(CMR)率均明显低于非缺失组(P值分别为0.034和0.036),且复发率明显高于非缺失组(P=0.033);p16基因缺失组使用伊马替尼联合化疗者和使用达沙替尼联合化疗者的MMR、CMR率及复发率差异均无统计学意义(P值均>0.05);p16基因缺失组患者3年总体生存(OS)率及无病生存(DFS)率分别为37.1%和12.4%,显著低于非缺失组的54.1%和45.9%(P值分别为0.037和0.026);25例p16基因缺失患者中14例行异基因造血干细胞移植(allo-HSCT),其中位OS时间为21个月,明显长于非移植组患者的12个月(P=0.030)。
成人Ph+ALL伴p16基因缺失患者预后相对较差,二代酪氨酸激酶抑制剂不能明显改善其疗效,但allo-HSCT能够改善部分患者的生存,明确p16基因缺失状态对于评估预后和指导临床治疗有重要意义。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Ph染色体是急性淋巴细胞白血病(ALL)常见的细胞遗传学异常,酪氨酸激酶抑制剂(TKI)联合化疗及异基因造血干细胞移植(allo-HSCT)可显著改善Ph+ALL的预后,但是仍有部分患者出现耐药、复发甚至疾病进展[1]。TKI耐药仍是目前Ph+ALL治疗的一大难点,因此,探讨TKI耐药机制以及寻找新的预后标志对于该部分患者显得尤其重要。染色体9p21的异常在多种肿瘤的发生发展中起重要作用[2]。目前已经发现在9p21区域有CDKN2A(p16)和CDKN2B(p15)两个重要的抑癌基因,它们分别编码p16(INK4a)/p14(ARF)和p15(INK4b)周期蛋白,构成了一个重要的抑癌基因簇[3]。在ALL中,18.2%~45.0%的患者可以检测到p16基因缺失,我们前期研究及目前文献报道p16基因缺失可能是ALL预后不良的独立危险因素[4,5]。本研究中我们应用FISH技术检测80例初诊Ph+ALL患者p16基因缺失情况,并进一步分析该基因缺失与Ph+ALL患者临床特征、疗效及预后关系。
对2010年1月至2015年4月就诊于我院血液科的80例初诊Ph+ALL患者的临床资料进行回顾性分析,所有患者均根据临床表现、骨髓细胞形态学、免疫分型、细胞遗传学及分子生物学结果进行诊断,符合WHO(2008)急性白血病诊断标准。骨髓细胞核型分析采用常规G显带法。骨髓细胞免疫表型分析采用流式细胞术检测淋系和髓系抗原。BCR-ABL mRNA检测采用常规实时荧光定量PCR(RQ-PCR)方法,以ABL作为内参基因,BCR-ABL mRNA水平以BCR-ABL/ABL基因转录本拷贝数×100%表示,敏感度为MR4.5(BCR-ABLIS=0.0032%)[6]。
针对p16基因缺失的预包被式探针诊断系统购于英国Cytocell公司,按设计组合标记固定于一张检测玻片,按试剂盒说明书进行操作。探针组包括:覆盖9p21区域101 kb的红色探针,9号染色体质控探针(D923,9q12上的异染色质块,绿色)。正常细胞的荧光原位杂交信号为2红2绿,1红2绿表明p16基因从1条染色体中缺失,即杂合性缺失,0红2绿为纯合性缺失(图1)。杂合性缺失与纯合性缺失均定义为缺失。根据ISCN(2005)标准分析检测结果,每份间期核标本计数200个细胞[7]。

A:正常细胞;B:杂合性缺失;C:纯合性缺失
所有Ph+ALL患者均首先予VILP(长春新碱、去甲氧柔红霉素、左旋门冬酰胺酶、泼尼松)或VDLP(长春新碱、柔红霉素、左旋门冬酰胺酶、泼尼松)方案进行诱导化疗,明确诊断后尽早联合TKI药物治疗(伊马替尼400 mg每日1次或达沙替尼100 mg每日1次)。诱导缓解后予Hyper CVAD-A(环磷酰胺、长春新碱、阿霉素、地塞米松)方案和Hyper CVAD-B(大剂量甲氨蝶呤和阿糖胞苷)方案交替巩固治疗。其中54例患者在巩固化疗2~5个疗程后进行allo-HSCT。对所有患者均进行腰椎穿刺并鞘内注射药物(阿糖胞苷+甲氨蝶呤+地塞米松)预防中枢神经系统白血病。
首疗程诱导治疗结束后行骨髓细胞形态学检查,评价血液学缓解情况;3个疗程后疗效评价主要根据细胞分子学反应。完全缓解(CR)定义为化疗后患者骨髓中原始细胞≤0.050,外周血中性粒细胞绝对计数≥1.5×109/L,PLT≥100×109/L,白细胞分类中无白血病细胞,无髓外白血病。主要分子学反应(MMR)定义为BCR-ABL mRNA较治疗前下降至少3个对数级(BCR-ABL/ABL≤0.1%)。完全分子学反应(CMR)定义为BCR-ABL mRNA拷贝数低于检测极限水平(BCR-ABL/ABL≤0.0032%)[6,8]。复发定义为获得CR患者骨髓中原始细胞≥0.050或者发生髓外侵犯。总生存(OS)时间指从诊断起至患者死亡(包括任何原因)或末次随访日止。无病生存(DFS)时间定义为自CR至白血病复发或在CR期间死亡或末次随访日。随访截止日期为2016年5月4日。
应用SPSS 20.0统计学软件进行分析,样本均数的比较采用t检验,率的比较采用卡方检验,生存率的比较采用Log-rank检验,采用Kaplan-Meier法绘制生存曲线。P<0.05为差异有统计学意义。
本组80例成人Ph+ALL患者中位年龄33.5(18~61)岁。25例(31.3%)患者检出p16基因缺失,其中1例为杂合缺失,24例为纯合缺失。p16基因缺失与未缺失组相比,性别、年龄、中位WBC、骨髓中原始细胞比例、肝脾肿大及髓外浸润情况等差异均无统计学意义(P值均>0.05)。p16基因缺失患者初发高白细胞(WBC≥30×109/L)比非缺失组更常见(P=0.042)(表1)。在p16基因缺失组中,除外2例核型分析失败的患者,余23例中18例(78.3%)初诊核型分析存在Ph染色体伴附加染色体异常,发生率较非缺失组高(P=0.035),其中以累及7号染色体(4例,3例-7,1例7q-)、8号染色体(4例,3例易位)、19号染色体(3例,2例-19,1例+19)和der(22)(4例)更为常见。此外,本组病例中发现的Ph染色体变异型易位有t(8;9;22)(q21;q34;q11)、t(4;9;22)(q21;q34;q11)、t(9;9;22)(q22;p24;q11)。
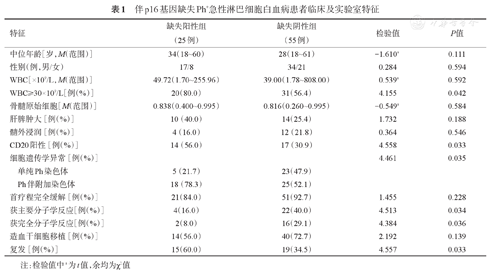
伴p16基因缺失Ph+急性淋巴细胞白血病患者临床及实验室特征
伴p16基因缺失Ph+急性淋巴细胞白血病患者临床及实验室特征
| 特征 | 缺失阳性组(25例) | 缺失阴性组(55例) | 检验值 | P值 | |
|---|---|---|---|---|---|
| 中位年龄[岁,M(范围)] | 34(18~60) | 28(18~61) | -1.610a | 0.111 | |
| 性别(例,男/女) | 17/8 | 34/21 | 0.284 | 0.594 | |
| WBC[×109/L,M(范围)] | 49.72(1.70~255.96) | 39.00(1.78~808.00) | 0.539a | 0.592 | |
| WBC≥30×109/L[例(%)] | 20(80.0) | 31(56.4) | 4.155 | 0.042 | |
| 骨髓原始细胞[M(范围)] | 0.838(0.400~0.995) | 0.816(0.260~0.995) | -0.549a | 0.584 | |
| 肝脾肿大[例(%)] | 10(40.0) | 14(25.4) | 1.732 | 0.188 | |
| 髓外浸润[例(%)] | 4(16.0) | 12(21.8) | 0.364 | 0.546 | |
| CD20阳性[例(%)] | 14(56.0) | 17(30.9) | 4.558 | 0.033 | |
| 细胞遗传学异常[例(%)] | 4.461 | 0.035 | |||
| 单纯Ph染色体 | 5(21.7) | 23(47.9) | |||
| Ph伴附加染色体 | 18(78.3) | 25(52.1) | |||
| 首疗程完全缓解[例(%)] | 21(84.0) | 51(92.7) | 1.455 | 0.228 | |
| 获主要分子学反应[例(%)] | 4(16.0) | 22(40.0) | 4.513 | 0.034 | |
| 获完全分子学反应[例(%)] | 2(8.0) | 16(29.1) | 4.384 | 0.036 | |
| 造血干细胞移植[例(%)] | 14(56.0) | 40(72.7) | 2.192 | 0.139 | |
| 复发[例(%)] | 15(60.0) | 19(34.5) | 4.557 | 0.033 | |
注:检验值中a为t值,余均为χ2值
对80例成人Ph+ALL患者骨髓细胞进行免疫表型分析,CD抗原表达>20%为阳性。p16基因缺失与未缺失组相比,除CD20外,两组间CD45、CD13、CD14、CD15、CD117、CD33、CD34、CD38、HLA-DR、CD10、CD19、CD22、CD2、CD3、CD7、CD11b、CD56等表达差异均无统计学意义(P值均>0.05),p16基因缺失患者多合并CD20表达(P=0.033)。
p16基因缺失组使用VDLP诱导方案14例,VILP诱导方案11例,非缺失组分别为37例和18例,两组差异无统计学意义(P=0.331)。p16基因缺失组与非缺失组患者化疗联合伊马替尼治疗分别为13例和29例,联合达沙替尼治疗分别为12例和26例,两组差异无统计学意义(P=0.952)。p16基因缺失组与非缺失组相比,首疗程诱导治疗CR率差异无统计学意义(84.0%对92.7%,P=0.249)。p16基因缺失组治疗3个疗程后MMR率为16.0%,CMR率为8.0%,显著低于非缺失组的40.0%和29.1%(P值分别为0.034和0.036)(表1)。两组患者BCR-ABL转录本均以P190为主,p16基因缺失组中P190阳性19例,p210阳性6例,非缺失组分别为40例和15例,差异无统计学意义(P=0.758)。p16基因缺失组中15例(60.0%)患者出现复发(血液学或中枢神经系统复发),复发率明显高于非缺失组(34.5%,P=0.033)。p16基因缺失组患者中化疗联合伊马替尼治疗13例,联合达沙替尼治疗12例,两者的MMR(15.4%对16.7%,P=0.930)、CMR率(7.7%对8.3%,P=0.953)及复发率(61.5%对58.3%,P=0.870)差异均无统计学意义。
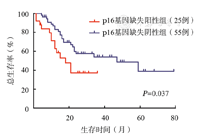
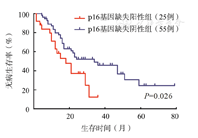
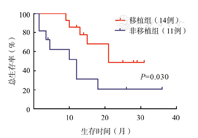
80例成人Ph+ALL患者中54例在巩固化疗2~5个疗程后进行allo-HSCT,其中p16基因缺失组14例,非缺失组40例,截至末次随访,80例患者中位随访时间为19.5(2~79)个月。p16基因缺失组和非缺失组患者中位生存时间分别为18和47个月(P=0.037)。p16基因缺失组患者3年OS率及DFS率分别为37.1%、12.4%,而非缺失组分别为54.1%、45.9%,差异均有统计学意义(P值分别为0.026和0.030)(图2、图3)。p16基因缺失患者中移植组中位生存时间为21个月,非移植组为12个月(P=0.030)(图4)。
2014年NCCN指南中将ALL分为高危组和标危组,定义高危组ALL的危险因素有:①Ph阳性或BCR-ABL基因阳性;②t(v;11q23)或MLL基因重排;③诊断时WBC≥30×109/L(B-ALL),≥100×109/L(T-ALL);④低二倍体(染色体数<44条)。标危组ALL则不具有上述任何危险因素之一[7]。近年基于疾病危险度的分层诊断和TKI的应用及allo-HSCT方案的优化显著改善了Ph+ALL患者的预后,但是仍有部分患者出现复发,难以从目前的诊疗中获益,强烈提示此类疾病需要再分层。我们曾报道p16基因缺失儿童和成人B-ALL患者预后相对较差[4, 9],且国外有报道,p16基因缺失是Ph+ALL除BCR-ABL融合基因外最常见的基因突变之一,其检出率为29.2%~64.0%[10,11]。Mullighan等[12]还发现Ph+ALL和慢性髓性白血病ALL变患者伴p16基因缺失的细胞遗传学改变可能是影响伊马替尼疗效的原因之一。
本研究结果显示,p16基因缺失在成人Ph+ALL中的发生率为31.3%,与文献[9、10、11]报道相符。我们发现,p16基因缺失组患者具有初诊高白细胞(WBC≥30×109/L)、CD20阳性率高以及伴随附加染色体异常更常见的特点。目前CD20阳性对Ph+ ALL预后的影响仍然存在争议[13,14],但是多数研究认为CD20阳性与ALL复发率增高、CR持续时间缩短及生存率降低密切相关[15,16],利妥昔单抗联合标准化疗能有效延长60岁以下CD20阳性ALL患者的OS时间[16]。基于上述研究及我们发现p16基因缺失组患者更容易高表达CD20、预后差、复发率高等特点,我们研究组推测利妥昔单抗有可能改善p16基因缺失患者的预后,但目前无相关报道,仍需进一步研究证实。本研究结果还显示,p16基因缺失组患者更易伴随附加染色体异常,可能主要因为p16基因可以通过抑制CDK4/6-cyclinD复合物激酶活性,使Rb呈持续高磷酸的活化状态,从而抑制细胞由G1期进入到S期,导致细胞周期停滞[3,17]。本研究中p16基因缺失患者出现附加染色体异常以累及7、8、19号染色体以及der(22)更为常见,Wetzler等[18]的研究亦表明Ph+ALL患者伴7号染色体异常和der(22)异常往往提示预后不良。
我们在研究中发现,Ph+ ALL伴p16基因缺失患者治疗效果差,复发率高,3年OS及DFS率明显低于非缺失患者,与国外大部分报道一致[19,20,21]。p16基因缺失组患者中化疗联合伊马替尼治疗13例,联合达沙替尼治疗12例,两者的MMR、CMR、复发率差异无统计学意义。早有报道伊马替尼治疗p16基因缺失Ph+ALL的效果不佳[12],Williams等[22,23]通过用BCR-ABL基因转染p16基因缺失的小鼠骨髓细胞发现,该小鼠对伊马替尼治疗敏感性差,并且证实p16基因缺失通过细胞因子IL-7依赖形式诱导细胞对伊马替尼耐药。目前越来越多的报道显示,二代TKI(达沙替尼)联合化疗可明显改善Ph+ ALL疗效及预后[24,25],国外多中心研究发现仅接受达沙替尼联合化疗治疗的Ph+ALL患者可获得持续缓解和长期生存(中位OS时间为55个月)[24]。而本研究发现,Ph+ALL伴p16基因缺失患者应用二代TKI如达沙替尼并不能获得更深层次的MMR以及减少复发,也许与本研究病例数少有关。目前尚缺少针对p16基因缺失的靶向治疗药物,但我们的研究结果显示,allo-HSCT能够改善伴p16基因缺失患者的预后,为该部分患者的治疗选择提供了数据。
综上所述,成人Ph+ALL伴p16基因缺失的患者预后相对较差,使用二代TKI不能明显改善该部分患者的疗效,但allo-HSCT能够延长患者的生存时间,因此,明确p16基因缺失状态对于Ph+ALL预后评估和指导临床治疗具有重要意义。

























