
探讨不明原因智力障碍共患癫痫(ID-E)患者的临床特点、病因及预后。
对2015年3月至2016年3月中南大学湘雅医院儿科40例不明原因ID-E患者的临床特点、病因及癫痫控制情况进行回顾性分析,并对所有患者进行随访。
40例不明原因ID-E患者中男25例(62.5%),重度智力障碍34例(85.0%)。癫痫首次发作年龄为0.16~8.00岁(中位年龄1.5岁),脑电图背景慢化20例(50.0%),局灶性放电22例(55.0%)。头颅磁共振异常10例(25.0%),主要为脑发育异常及脑萎缩。随访时间0.58~1.58年,癫痫发作控制19例(47.5%),25例(62.5%)在病程中使用2种及2种以上抗癫痫药物,药物难治性癫痫患者占47.5%(19例)。癫痫发作控制患儿及未控制患儿精神运动发育改善分别为12例(63.2%)及2例(9.5%)。通过全基因组拷贝数变异(CNVs)检测及CNVs检测阴性部分患者进行基因靶向捕获测序检测,分别有8例(8/40例,20.0%)及3例(3/16例,18.8%)明确了遗传学病因。
不明原因ID-E患者男性多见,智力受损程度重,药物难治性癫痫比例高,有效控制癫痫发作有助于患者精神运动发育。遗传病因学检测对于癫痫的控制、预后评估有重要作用,全基因组CNVs检测阳性率高,可作为临床一线遗传学检测手段。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
智力障碍(intellectual disability,ID)指起病于发育阶段(或18岁之前)的一种功能性疾病状态,其特征为智力和适应能力受限,人群中的发病率为1%~3%[1]。ID可分为综合征型ID(syndromic ID,S-ID)及非综合征型ID(non-syndromic ID),其中S-ID表现为ID的同时存在一个或多个临床异常表现,如孤独症谱系障碍、癫痫、面部发育畸形或先天性异常等。在ID人群中,尤其是S-ID患者,慢性疾病的发生率较普通人群高,其中癫痫在所有慢性共患疾病中排首位[2],共患率为20%~30%,且随着ID程度加重而提高[3]。Mullen等[4]报道ID共患癫痫(ID-E)患者的基因拷贝数变异(CNVs)率明显高于ID组和遗传性癫痫组,提示ID-E患者有更强的遗传关联性,同时ID-E患者的癫痫发作控制率及病死率也明显高于单纯ID或单纯癫痫的患者[2],因此ID-E患者的临床特点、治疗等具有其特殊性及难度。
不明原因ID-E患者指排除非遗传因素、染色体核型异常(如21-三体综合征等)及明显的单基因病后病因不明的ID-E患者,推测不明原因的ID-E患者治疗、预后与遗传学病因有重要的相关性。目前,国际上针对不明原因ID患者推荐的一线遗传学检测为全基因组CNVs[5,6],因此,本研究通过对不明原因ID-E患者的临床资料进行分析,并随访其癫痫发作控制及智能改善情况,结合遗传学病因,探讨不明原因ID-E发病的临床特点、治疗和预后,以提高其生存质量。
2015年3月至2016年3月在中南大学湘雅医院儿科就诊的不明原因ID-E患儿40例,入组前均获得患儿监护人同意,并通过医院医学伦理委员会审查批准。
ID的诊断标准参照《精神疾病诊断与统计手册》第5版(DSM-Ⅴ)诊断[7]。ID程度分级依照IQ受损程度分为轻度(IQ:50~69)、重度(IQ:<50)。癫痫诊断依照临床表现及脑电图(EEG)等综合判断。
纳入标准:符合上述诊断标准的ID-E患者,ID或精神运动发育迟滞应在癫痫发作前,且排除明确的非遗传致病因素(如缺氧缺血性脑病、脑血管病变、外伤等)、染色体病(21-三体综合征等)、临床表型明确的单基因病。
40例患者均完成了19导联视频EEG监测及头颅MRI检查。
通过单核苷酸多态性芯片(single nucleotide polymorphism-based array,SNP-array)检测致病性CNVs。16例上述检测阴性患者后续进行了靶向捕获二代测序。
采用现场调查的方式收集详细临床资料,并进行小儿神经专科门诊随访。治疗效果评估标准为服用抗癫痫药物(AEDs)达目标剂量后其临床发作程度及频率变化情况,完全控制指无癫痫发作超过6个月,有效指临床发作减少≥50%,无效指临床发作未控制(发作减少<50%)或加重。治疗后智能评估依照智力测试或患儿家属对患儿精神运动发育改善情况的描述进行随访。
40例患儿中男25例(62.5%),女15例(37.5%);ID受损程度:轻度6例(15.0%),重度34例(85.0%);首次就诊原因:癫痫14例,全面发育迟缓11例,运动发育迟缓8例,语言发育迟缓3例,精神运动发育倒退3例,孤独症1例。
癫痫发病年龄为0.16~8.00(2.50±0.35)岁,中位年龄1.5岁;14例首发年龄<1岁,占35.0%。发作形式:局限发作25例,全面性强直阵挛发作7例,强直发作9例,痉挛发作4例,失神发作2例;9例患者有2种及2种以上发作形式;12例患者发作具有热敏感性,占30.0%。
EEG结果:背景活动慢化20例(50.0%);发作期监测到临床发作11例,分别为局限发作8例,痉挛发作1例,肌阵挛发作2例,失张力发作1例,不典型失神发作2例,全面性强直阵挛发作2例,其中5例监测到2种及2种以上发作形式;发作间期表现为局灶性或多灶性放电22例,广泛性放电15例,不典型高度失律3例。头颅MRI结果:正常30例,占75.0%;异常10例,占25.0%,其中脑白质异常7例,脑萎缩、胼胝体发育不良及巨脑回各1例。
40例患者均予以AEDs治疗,单药控制发作14例(1例为发作减少50%以上),25例使用或使用过2种及2种以上AEDs,占62.5%,其中控制发作6例(1例为发作减少50%以上),19例无效,药物难治性癫痫比例47.5%。使用左乙拉西坦(LEV)32例,单药控制率为66.6%(14/21例);使用丙戊酸钠20例,单药控制率为16.7%(2/12例);使用托吡酯12例,单药控制率25%(1/4例);使用奥卡西平6例,单药控制率为66.7%(2/3例);硝西泮添加治疗4例。同时,药物难治性癫痫患者中5例患者进行了生酮饮食,癫痫发作均未控制,3例精神状态较前好转。随访时间0.58~1.58年,癫痫发作控制19例(47.5%),无效21例(52.5%)。癫痫发作控制患儿,家长自觉患者精神运动发育改善9例(63.2%),倒退2例,无明显改善8例;癫痫发作未控制患儿,家长自觉患者精神运动发育改善2例(9.5%),倒退8例,无明显改善11例。
40例不明原因ID-E患者中,8例患者携带至少1个致病性或可疑致病性CNVs,阳性率为20.0%,缺失碱基长度0.22~7.00 Mb。1例患者检测出22q11微缺失,变现为轻度智力受损、癫痫、面容异常、先天性心脏病(房间隔缺损),符合典型的22q11微缺失综合征;1例检出4p16.1-3缺失6.5 Mb,重度ID、癫痫、宫内发育受限、典型"希腊战士头盔"面容(小头畸形、宽鼻翼等),符合Wolf-Hirschhorn综合征;1例检出17p11.2缺失3.4 Mb,表现为重度ID,行为异常(多动、注意力不集中),癫痫(8岁起病),生长发育明显迟缓(11岁:身高126 cm,体质量27.5 kg),四肢末端短粗,缺失片段包含Smith-Magenis综合征(SMS)关键基因RAI1,符合SMS;1例检测出14q12缺失0.8 Mb,包含致病基因FOXG1,FOXG1基因突变典型表现为获得性小头畸形、癫痫、运动认知发育迟缓、重度ID,此患者临床表现相符,提示为致病性的变异可能性大;1例检出5p14.3缺失1.46 Mb,DECIPHER数据库收录该片段缺失患者,其中3例表现为ID或语言学习功能受限(288889,289890,322791),1例表现为癫痫(Patient 270797),因患儿父亲已去世,故不能父母验证,考虑此突变为可能致病性CNVs;1例检出1q31.1缺失0.22 Mb,DECIPHER数据库报道此区域缺失表型为整体发育迟缓,父母尚未完善验证。16例CNVs检测阴性的患者后续进行了基因靶向捕获高通量测序,3例检测出致病突变并完善一代测序验证,阳性率18.8%。具体突变信息:2例男童检测出MECP2基因突变,1例为半合子移码突变(c.1200_1243del43),其母亲为携带者,智力稍差,有热性惊厥病史;另1例为错义突变(c.628T>C),此位点高度保守,SIFT、Polyphen、Mutation_Taster预测为有害性突变,人类基因突变数据库(HGMD)尚未报道此位点突变,突变源于母亲,高度考虑此2例患者为MECP2基因突变导致的综合征型精神发育迟滞。1例患者检测出KCNB1基因杂合错义突变(c.G1136T)p.G379V,此位点高度保守,SIFT、Polyphen、Mutation_Taster预测为有害性突变,HGMD尚未报道此位点突变,患者表型为ID共患难治性癫痫,考虑为KCNB1基因突变导致的癫痫脑病26型。CNVs检测阳性率为20.0%(8/40例),靶向捕获基因检测阳性率18.8%(3/16例),遗传学检测阴性及阳性患者临床资料见表1。
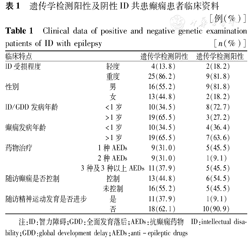
遗传学检测阳性及阴性ID共患癫痫患者临床资料 [例(%)]
Clinical data of positive and negative genetic examination patients of ID with epilepsy [n(%)]
遗传学检测阳性及阴性ID共患癫痫患者临床资料 [例(%)]
Clinical data of positive and negative genetic examination patients of ID with epilepsy [n(%)]
| 临床特点 | 遗传学检测阴性 | 遗传学检测阳性 | |
|---|---|---|---|
| ID受损程度 | 轻度 | 4(13.8) | 2(18.2) |
| 重度 | 25(86.2) | 9(81.8) | |
| 性别 | 男 | 16(55.2) | 9(81.8) |
| 女 | 13(44.8) | 2(18.2) | |
| ID/GDD发病年龄 | <1岁 | 10(34.5) | 8(72.7) |
| >1岁 | 19(65.5) | 3(27.2) | |
| 癫痫发病年龄 | <1岁 | 10(34.5) | 4(36.4) |
| >1岁 | 19(65.5) | 7(63.6) | |
| 药物治疗 | 1种AEDs | 9(31.0) | 5(45.5) |
| 2种AEDs | 9(31.0) | 1(9.1) | |
| 3种及3种以上AEDs | 11(37.9) | 5(45.5) | |
| 随访癫痫是否控制 | 控制 | 13(44.8) | 6(54.5) |
| 未控制 | 16(55.2) | 5(45.5) | |
| 随访精神运动发育是否进步 | 是 | 11(37.9) | 1(9.1) |
| 否 | 18(62.1) | 10(90.9) |
注:ID:智力障碍;GDD:全面发育落后;AEDs:抗癫痫药物 ID:intellectual disability;GDD:global development delay;AEDs:anti-epileptic drugs
癫痫作为ID患者中最常见的慢性共患病,ID-E患者的临床特点及病因一直是国内外的研究热点。因此,本研究主要针对不明原因ID-E患者进行临床及遗传学病因进行分析。40例患者中重度ID患者占85.0%。一篇关于ID儿童共患慢性疾病的系统综述中分析了来自不同国家或地区的14个相关研究,指出癫痫在ID患儿中的发病率为5.5%~35.0%,平均发病率为22.0%(95%CI:20.8%~23.2%)[2],明显高于普通人群。其中5.5%的癫痫最低发病率纳入的研究对象为边缘性ID或轻度智力受损,35%的癫痫最高发病率纳入的研究对象ID受损程度为中度至极重度,表明重度ID患者癫痫发病率更高。本组ID-E患者男童占62.5%,同国外报道的儿童ID-E患者中男性比例相仿[2],考虑男性ID患者可能更易合并癫痫发作,具体机制目前尚不明确。40例ID-E患者中,部分患者初次就诊病因为抽搐发作,因此对于不明原因抽搐发作的患者一定要详细询问抽搐前生长发育情况;其次,在ID患者,尤其重度ID患者,需告知家属病程中出现抽搐发作的可能性大,对于典型或不典型抽搐发作均应重视,并争取早期完善EEG、MRI等临床检查。
本组ID-E患者中癫痫发病年龄跨度广,无明显的年龄集中区段,但癫痫发作具有一定的特点:(1)局限性发作常见;(2)部分患者既往有高热抽搐病史或发作具有热敏感性;(3)病程中使用2种或2种以上AEDs患者所占比例高(62.5%),癫痫的控制率低,且药物难治性癫痫占有比例高(47.5%)。40例患者中使用最多的AEDs是LEV,其次为丙戊酸、托吡酯、奥卡西平。Doran等[8]在2016年对ID-E患者AEDs选择的分级证据进行了系统研究,对43篇相关文献进行回顾性分析,并未得出具体某种AEDs的治疗效果最佳或不良反应最小,因此尚无药物推荐指南。但文献中指出新型AEDs较传统AEDs耐受性好,认知功能损害低。LEV在本组患者中的应用较多,单药控制率达66.6%。Barca等[9]指出LEV应作为21-三体综合征合并全面性癫痫发作患者的一线推荐治疗。张舒岩等[10]指出LEV单药治疗ID-E儿童患者,可以减轻患者的认知功能损害,提高反应能力。另外,苯二氮艹,卓)类药物在ID-E患者中也广泛使用,尤其针对药物难治性ID-E患者[8]。氯巴占被认为是一种安全有效的添加治疗药物,在国外广泛使用[11],本组患者中有4例难治性癫痫使用了苯二氮艹),\s\do5(卓))类,国内因暂时没有氯巴占药物来源,故使用均为硝西泮。
癫痫的发作特点及预后与遗传学病因有重要的关系。Bao等[12]针对共患癫痫发作的Rett综合征患者进行了MECP2基因突变位点与癫痫药物控制率相关性研究,发现MECP2基因的p.R106W、p.R168X及p.R255X突变位点的患者更易出现药物难治性癫痫。不明原因ID-E患者考虑其有潜在的遗传致病因素,因此,遗传学病因筛查具有重要的临床意义。本组ID-E患者均完成了全基因组CNVs检测,阳性率为20.0%,高于ID/全面发育落后(GDD)患者的CNVs检出率(14%)[13],低于文献报道的遗传性癫痫共患ID的检出率(28%)[4]。涉及区域有15q11-13、22q11.21、4p16.1-3、17p11.2、5p14.3、14q12、1q31.1等。其中包含易共患癫痫发作的S-ID,如15q11-13缺失引起的快乐木偶综合征,重要的表型为ID-E发作[14]。同时,也包括罕见的微缺失综合征,如17p11.2缺失引起的Smith-Magenis综合征,发病率仅为125 000~115 000[15]。CNVs检测阳性患者中S-ID患者比例高,这是因为ID-E患者在表现为ID同时共患癫痫,且部分患者有面容异常、心脏发育异常、皮肤异常等其他系统受累,故表现为S-ID概率高。因此,对于ID-E患者,需要详细全面的体格检查(细微的面容异常,指端异常,如第5指微弯曲),并完善心脏彩超、腹部彩超等检查,防止遗漏可能存在的其他系统异常。全基因组CNVs检测阴性的患者有16例后续完善靶向捕获测序,检出3例携带致病性突变,阳性率为18.8%。其中KCNB1基因突变患者为国内首次报道[16],且突变位点为新突变位点,国外已报道的KCNB1突变患儿均共患药物难治性癫痫[17],故告知患者父母预后差,并为其提供相关遗传咨询。40例ID-E患者的遗传学检测总阳性率为38.8%,低于相关文献报道[18,19],考虑原因可能是本研究排除了染色体核型分析异常的患者(整条染色体重复或缺失、大片段异位倒位等),同时有部分CNVs阴性患者因家庭经济原因,尚未进行Panel检测,造成可能存在的选择偏倚,其次样本量小。
综上所述,不明原因ID-E患者临床特点、发作形式、抗癫痫治疗具有其特殊性,同时遗传学病因学检测对于患者癫痫控制、预后评估有重要作用。因此,后续需进行大样本量、更为深入、全面的遗传学筛查,并将遗传学结果与临床表型相结合,探索ID-E发作的根本发病机制,改善患者预后。

























