
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
1932年Cushing教授就指出颅咽管瘤是最困扰神经外科医生的颅内肿瘤。直到今天,对颅咽管瘤患者实施较为满意的手术治疗仍然是神经外科医师所面临的巨大挑战。一方面目前对于颅咽管瘤最佳治疗策略仍存在争论,另一方面由于对颅咽管瘤的认识参差不齐,不同医疗机构对颅咽管瘤的处理方案差异很大。此外,对于术后水电解质紊乱、内分泌功能障碍、肥胖、认知功能障碍等垂体-下丘脑功能障碍的认识不足,严重影响颅咽管瘤患者的预后。为了提高颅咽管瘤治疗水平,中华医学会神经外科学分会小儿神经外科学组,组织了神经外科、放疗科及内分泌科等有关专业的专家们撰写了《颅咽管瘤治疗专家共识》(以下简称本共识),希望通过本共识使我国医师对颅咽管瘤病理生理状态加深理解,提高颅咽管瘤治疗的水平,规范治疗,进而改善患者的预后。
颅咽管瘤占颅内肿瘤的2%~5%,是儿童常见的颅内肿瘤,位居儿童颅内肿瘤的第2位,约占儿童颅内肿瘤的5.6%~15%,占儿童鞍区肿瘤的54%[1]。颅咽管瘤主要有2个发病高峰期:5~15岁的儿童以及40岁左右的成人[2]。发病具有明显的地域特征,多见于东亚地区和非洲。
手术是治疗颅咽管瘤的主要手段。颅咽管瘤虽然是良性肿瘤,但位置深在,毗邻重要神经血管结构,部分患者手术难度较高。因此,三维适形放疗、立体定向放疗、ommaya囊植入并核素内照射、化疗等治疗方式仍然在广泛使用。颅咽管瘤的手术治疗需要在充分保护垂体-下丘脑功能及视路结构的前提下积极地追求全切除方能治愈。对于严重侵犯下丘脑结构的颅咽管瘤,手术中对第三脑室壁结构与肿瘤间形态学关系的充分辨识至关重要,可最大限度地提高其全切除率。如果术后肿瘤残留,在充分告知患者放疗并发症(导致内分泌水平下降、导致再次手术困难等)的前提下,放疗是可能的选择[3]。因为治疗策略与手术水平的不一,因此,不同报道的复发率、生存率、垂体-下丘脑功能、生存质量等差别较大[4,5,6]。
颅咽管瘤是生长相对缓慢的脑外肿瘤,除内分泌症状外,通常在肿瘤较大的时候才开始出现临床表现。主要为:颅内压增高症状(头痛、恶心、呕吐)、视力下降、垂体-下丘脑功能障碍。当肿瘤发展到一定程度,这几种临床表现可以同时存在。儿童患者的生长发育和视力障碍发生率较高,成人内分泌功能障碍发生率较高。内分泌功能障碍包括生长激素(GH)、卵泡刺激素(FSH)、黄体生成素(LH)、促肾上腺皮质激素(ACTH)、促甲状腺激素(TSH)缺乏[7]。部分颅咽管瘤患者具有下丘脑功能障碍,主要为:肥胖,疲劳,行为变化,昼夜睡眠节律不规律,渴感消失,体温、心率和血压变化;甚至致死性下丘脑功能障碍[8]。患者出现视力下降和视野缺损的情况的发生率较高[9,10]。
颅咽管瘤位于鞍区,可向各个方向生长,个体差异较大。影像学上呈类圆形或分叶状。肿瘤为囊性、实性、混合性。囊液的磁共振成像(MRI)表现:T2WI大多数为高信号,部分为低信号(有角蛋白或钙盐结晶),T1WI因其成分不同而表现为低信号(含正铁血红蛋白)或高信号(其他蛋白含量高)。计算机断层扫描(CT):囊液在CT上多为低密度影。实性成分在CT上为不均匀、等或稍高密度。MRI的T1WI上信号强度与灰质相似;T2WI多为不均匀的高信号。实性部分及囊壁的MRI增强扫描可有明显或不均匀强化。
颅咽管瘤是常见的脑实质外起源的肿瘤,病理上分为成釉上皮型和鳞状乳头型[11]。目前研究认为,成釉上皮型颅咽管瘤主要是由残存于Rathke囊的上皮细胞CTNNB1基因外显子3发生突变,导致其编码的β-catenin不能被降解,激活经典wnt通路导致肿瘤发生[12,13,14];鳞状乳头型颅咽管瘤可能是由于残存于结节漏斗部的Rathke囊的上皮细胞发生鳞状化生导致,近年来BRAF V600E突变被发现广泛存在于鳞状乳头型颅咽管瘤中[15]。成釉上皮型颅咽管瘤主要以指轮状细胞、栅栏样上皮细胞、星网状细胞、湿性角化物以及散在的钙化为主要病理学特征,与神经组织毗邻处往往可见到胶质增生带,部分与下丘脑毗邻处可见肿瘤呈指状生长进入下丘脑神经组织内[16],但不属于三脑室内肿瘤。鳞状乳头型颅咽管瘤以复层鳞状上皮形成乳头状为主要特征,乳头结构中心可见血管,钙化、湿性角化物罕见。两型颅咽管瘤间质中均可见到不同程度的炎症细胞浸润。既往文献还报道,混合型颅咽管瘤,即同时拥有两种类型颅咽管瘤的病理学特征,现在认为混合型颅咽管瘤并不存在[17]。
国内外学者根据解剖位置、与视交叉关系、对三脑室底推挤的程度等对颅咽管瘤进行分型。例如Yasargil等[22]、Wang等[23]及Steno等[24]使用的鞍下或鞍上型,脑室内或脑室外型肿瘤;Hoffman[25]使用的视交叉前或视交叉后型肿瘤;Kitano等[26]使用的视交叉下型肿瘤;Kassam等[27]使用的漏斗部前、穿漏斗部和漏斗部后型肿瘤;以及一些学者基于起源位置和周边膜性结构关系提出的QST分型[28,29,30]。肿瘤的分型可以解释肿瘤的位置以及生长模式,能为手术入路的选取提供帮助。颅咽管瘤是一种起源于脑实质外的肿瘤,但是会凸入脑实质生长。颅外入路(经蝶、扩大经蝶等)、经颅入路(经翼点、扩大翼点等)、经颅经脑入路(经终板、胼胝体、侧脑室等)均被用于肿瘤切除。手术医生应该根据不同分型的颅咽管瘤,在不同入路的优势和使用不同入路的代价之间进行权衡,选择最佳预后的入路。Q型起源于鞍膈下,为鞍内、鞍内鞍上型,可以经颅或经蝶进行手术。S型起源于垂体柄蛛网膜袖套内,为鞍上脑室外型,可经颅或经蝶进行手术。T型起源于结节漏斗部,为结节漏斗型,建议选择经颅或经颅经脑入路,部分可选择经碟入路。对于侵入脑实质较多的肿瘤,尤其是高度超过中间块或超过前交通动脉1 cm的,建议选择经颅经脑入路。复发的患者或接受过放疗的患者,建议采取经颅联合入路。
颅咽管瘤切除术的关键是肿瘤与下丘脑-垂体柄及下丘脑组织之间关系的明确与辨识。肿瘤与颅内正常结构之间存在蛛网膜、软脑膜以及胶质反应层界面。在这些界面分离肿瘤不容易损伤正常神经组织及Willis环的细小分支血管。肿瘤的钙化需要经过仔细的锐性分离,多数情况下只要在直视下锐性分离就能安全地全切除。尽量识别和保留垂体柄,垂体柄的保留程度直接影响到术后内分泌紊乱的发生率和严重程度,术中垂体柄的辨认与保护可以作为下丘脑保护的标志性结构,应积极寻找和保护[22]。根据术前影像学表现判断垂体柄的位置,术中根据垂体柄与不同类型肿瘤的关系,尽可能多地或完整地保留垂体柄,可减少和减轻术后尿崩症[31]。

颅咽管瘤患者的围手术期管理流程
颅咽管瘤患者的围手术期管理流程
| 时间 | 检查项目 | |
|---|---|---|
| 术前 | ||
| 影像学:头颅CT、鞍区增强MRI | ||
| 垂体前叶:皮质醇(上午8:00采血,必要时行激发试验明确诊断);FT3、FT4、TSH(补充糖皮质激素后行甲状腺激素替代治疗);ACTH;GH、IGF-1;FSH、LH、T、E2、P、PRL | ||
| 垂体后叶:24 h尿量、尿比重、尿渗透压;尿崩患者必要时行去氨加压素试验 | ||
| 其他检查:视力、视野及眼底;身高、体质量及发育情况;常规术前检查 | ||
| 术后 | ||
| 1~3 d | 每日血生化、电解质(必要时增加监测频率);每小时尿量;24 h出入量、尿量、尿钠;中心静脉压;垂体前叶激素 | |
| 3~5 d | 每日血生化、电解质(必要时增加监测频率);每小时尿量(必要时);24 h出入量、尿量、尿钠;垂体前叶激素 | |
| 5~7 d | 每日血生化、电解质(必要时增加监测频率);24 h尿量、尿钠;垂体前叶激素 | |
注:FT3.游离三碘甲状腺原氨酸,FT4.游离甲状腺素,TSH.促甲状腺激素,ACTH.促肾上腺皮质激素,GH.生长激素,IGF-1.胰岛素样生长因子,FSH.卵泡刺激素,LH.黄体生成激素,T.睾酮,E2.雌二醇,P.孕激素,PRL.泌乳素
颅咽管瘤这样一个生长方式复杂多变的肿瘤,当临床考虑颅咽管瘤的诊断时,除常规检查外,需要完善以下检查:(1)鞍区平扫加增强MRI,鞍区三维CT平扫。必要时行CTA,MRA或数字减影血管造影(DSA)检查,评价肿瘤与血管的关系。(2)垂体前叶激素水平测定:皮质醇(F,上午8:00采血),测定促肾上腺皮质激素(ACTH)水平、甲状腺功能[游离三碘甲状腺原氨酸(FT3)、游离甲状腺素(FT4)、促甲状腺激素(TSH)]、生长激素(GH)水平、胰岛素样生长因子(IGF)-1水平、性激素6项[卵泡刺激素(FSH)、黄体生成激素(LH)、睾酮(T)、雌二醇(E2)、孕激素(P)及泌乳素(PRL)]、24 h尿游离皮质醇,清晨皮质醇为3~18 mg/L时需要进行ACTH激发实验。(3)多饮多尿症状明显患者:监测血浆渗透压,24 h尿量,24 h尿游离皮质醇,尿比重、尿渗透压及尿电解质情况。对于确诊中枢性尿崩困难的患者,应行加压素试验,以明确是否存在中枢性尿崩症。行血离子检查(钾钠氯)。(4)视力、视野检查:鞍区病变的占位压迫和手术所致的损伤,均可导致视神经功能暂时或永久受损。术前获得患者视力、视野情况,便于与术后视力、视野情况对比,有利于术中对视力、视野的保护。
颅咽管瘤术后发生尿崩比例较高,同时满足以下两个条件即可诊断尿崩:(1)血浆渗透压>300 mOsm/L,同时尿渗透压<300 mOsm/L,或者尿渗透压/血浆渗透压<1;(2)连续2 h尿量>4~5 ml·kg-1·h-1。典型过程分为3个阶段[32]:术后尿崩期(术后1~3 d左右),低血钠期(术后3~9 d左右),长期尿崩期(术后7~9 d之后)。应该在严密监测出入量和电解质的前提下,及时调整输入量以及输入液体的电解质比例,保持患者在手术后急性期内基本的水电解质平衡状态。轻、中度尿崩症,建议垂体后叶素肌注或口服去氨加压素治疗;重度尿崩症,建议使用去氨加压素或垂体后叶素持续微量泵注入,并监测中心静脉压。
对于高钠血症,限制钠盐和含钠液体输入;动态监测血钠水平,如果血钠水平继续上升,可以胃管定期注入白开水,并注意糖皮质激素的补充,必要时血液滤过;注意如果开始限尿治疗,谨慎使用降血钠治疗,防止血钠水平迅速下降导致严重后果。同时应监测血糖水平,若存在血糖升高,加重患者高渗状态,可以泵入胰岛素降糖。对于严重低钠血症,第1小时3%NaCl 150 ml静脉滴注20 min,20 min后复测血钠,目标1 h血钠上升5 mmol/L。1 h后症状无改善:继续静脉滴注3% NaCl,使血钠上升1 mmol/L/h,直到血钠达到130~135 mmol/h和症状改善。如1 h后症状改善根据尿量和尿钠的排出情况,继续静脉滴注3% NaCl。原则上第1个24 h内限制血钠上升在10 mmol/L,随后每日血钠上升<8 mmol/L,达到目标血钠130~135 mmol/L。但对于急性重度低钠血症,应尽快达到目标血钠。需要注意的是,过度快速纠正低钠血症可引起渗透性脱髓鞘综合征(ODS),建议补钠液浓度不超过3%。
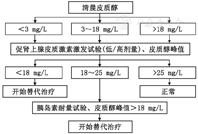
颅咽管瘤围手术期应该重点关注糖皮质激素的应用,术前应该根据皮质醇检测结果决定是否进行替代治疗(图1)。手术当天可予持续静脉输注氢化可的松,剂量为200~300 mg。术后1~3 d:术后监测尿量和电解质水平:如血钠偏高,在补液同时,可予小剂量(0.025~0.05 mg)去氨加压素(弥凝)对症治疗,儿童应注意根据其公斤体重进行剂量的调整剂量。术后第3~5天:根据患者的一般状态、食欲、血压、血钠,决定补充糖皮质激素剂量。静脉输注氢化可的松50~100 mg 2次/d;继续监测电解质和尿量,开始规律服用弥凝(根据尿量及体重调整剂量)。术后第5~7天:逐渐减少糖皮质激素剂量到氢化可的松20 mg 3次/d,或泼尼松5 mg 3次/d,根据患者病情,开始甲状腺激素替代治疗。
由于颅咽管瘤常常累及三脑室前部,周边下丘脑等结构功能重要,部分肿瘤与周边重要结构关系密切,为保护重要结构,可能导致部分肿瘤残存,这部分患者近期复发率高达50%,远期复发难免。因此,放射治疗(三维适形分割放疗、立体定向放疗和放射外科治疗)、囊内近距离放、化疗(P32/I131以及博来霉素等)、干扰素治疗等可作为延缓复发的治疗手段。但是这些姑息性治疗方法的远期疗效仍需进一步研究与评价。
放疗可延缓颅咽管瘤的复发,短期内控制肿瘤具有一定的疗效。对于多次复发、不能根治、年龄较大或难以耐受手术的患者可进行放射治疗。放射治疗可能引起肿瘤周围的下丘脑、视交叉、腺垂体、垂体柄等相邻部位及额叶的损伤,故治疗后患者可出现垂体功能低下表现以及记忆力减退等症状[31,33,34],同时会加重肿瘤与周围组织结构粘连,给再次手术带来困难。儿童颅咽管瘤患者,尤其是<6岁的患者,要尽量避免接受放射治疗,以减少对智力和内分泌方面的影响。对于不愿接受手术治疗或不能耐受手术的成人患者,在充分告知患者及家属放疗不良反应的情况下,放疗可以作为一种延长生存期的治疗手段。
Ommaya囊置入并同位素放疗是一种治疗囊性颅咽管瘤的方法,对于一些不愿意接受手术的儿童患者,可以通过置入Ommaya囊来推迟接受手术治疗[35]。接受Ommaya囊植入并不影响远期的预后,但可以有效推迟手术时间。要注意的是囊液会刺激周围组织形成肉芽,这样会导致引流管的各个洞口周围有很多组织包绕引流管生长,有的甚至长入引流管的引流口内,加大了手术切除的难度,术者需要特别注意这种情况[36]。
颅咽管瘤存在内分泌障碍是普遍现象,儿童患者的生长发育问题更加复杂,应该重视术后的长期激素替代治疗及随访。
对于轻度尿崩症患者,无需药物处理;对于中重度尿崩症患者,在补充体液丢失量的同时应给予抗利尿激素(ADH)治疗,控制尿量在200 ml/h左右。长期过量不恰当使用ADH药物会导致稀释性低钠血症,应注意定期复查血电解质。术后1个月内每周检查血电解质水平。术后1~6个月每个月检查电解质和肌酐水平(必要时加强监测频率)。根据血浆渗透压和血钠浓度以调整合适的剂量和给药间隔时间。部分低钠血症可通过补充糖皮质激素进行治疗,可经验性使用氢化可的松(50~100 mg/8 h,静脉给药),逐渐调整剂量到15~25 mg/d[37]。
对于肾上腺皮质激素分泌不足的患者首选氢化可的松进行替代治疗,15~25 mg,2~3次/d,也可应用泼尼松[38]。儿童用药剂量为6~10 mg·m-2·d-1,分2~3次服药。应该使用最小剂量的糖皮质激素模拟皮质醇生理分泌节律进行用药,50%~60%剂量在白天给药,使患者皮质醇水平达到正常值[39]。剂量调整主要依据临床经验及调整后患者是否出现新发或症状缓解,不合理的提升糖皮质激素剂量也容易导致肾上腺危象的发生[38]。
建议对甲状腺激素缺乏的患者使用左旋甲状腺素(L-T4)治疗,从低剂量开始逐渐增至25 μg/1~2周,儿童应根据其公斤体质量进行剂量的调整[40]。应先排除中枢性肾上腺低能症后再使用L-T4,以免出现肾上腺危象。如果在未评估肾上腺功能时开展了L-T4治疗,可预防性使用类固醇激素(氢化可的松或醋酸可的松)。治疗过程中需1~2个月调整一次剂量,使FT4逐渐升高到正常范围的中值水平。不应根据TSH水平调整药量[41,42]。
如果肿瘤术后1~2年,无复发迹象,可考虑生长激素替代治疗,生理剂量的生长激素,不会促进肿瘤复发[43]。对儿童和成人,补充生长激素都具有重要的意义。生长激素长期替代治疗过程中,应定期复查鞍区MRI。对于骨骺未闭合的儿童,生理剂量(0.1 U/kg)或更小剂量的生长激素,有助于身高增加,同时改善机体物质代谢,减少腹部脂肪,治疗效果良好。治疗期间,应监测身高增长幅度、甲状腺激素、血糖、IGF-1水平和骨龄。在替代治疗的过程中,甲状腺激素的剂量往往需要增加。IGF-1的水平升高到相应生物年龄(最好是骨龄)阶段的正常值范围内为宜。成人生长激素缺乏症的替代治疗应当遵循个体化原则,而不是根据体重决定剂量。建议从小剂量人重组生长激素开始(0.2 mg,睡前用),逐渐增加剂量,当恢复正常IGF-1值或出现疑似不良反应症状或临床症状改善(如:体脂分布、运动能力、神经心理表现、骨密度恢复,心血管事件危险因素减少)时停止增加剂量。<30岁的患者起始剂量要相应提高,而>60岁的患者起始剂量应控制在<0.2~0.4 mg/d。此外,对于性腺轴正常或口服雌激素或绝经后接受雌激素治疗的女性,生长激素替代剂量应适当提高。替代治疗目标为维持血浆IGF-1水平在相应年龄的正常范围内中上水平,剂量调整期每1~2个月复查,以后每6个月复查1次[44]。
暂时无生育需求的成年患者,应给予长期性激素替代治疗,以维持第二性征、增加骨密度。为推迟儿童患者骨骺闭合而获得更好的最终身高,应该在女童12~13岁,男童14~15岁开始少量性激素补充。对于成年男性患者,在除外禁忌证(红细胞增多症、严重睡眠呼吸暂停、前列腺癌)后,应根据年龄,症状和可能的合并症调整睾酮剂量,使血浆睾酮水平尽量接近正常值。可选择的药物有:十一酸睾酮口服制剂40~80 mg,3次/d或长效十一酸睾酮注射制剂250 mg肌内注射,1次/月。睾酮替代治疗期间,应通过检测男性胡须生长,肌肉质量及力量,血红蛋白,红细胞计数及血细胞比容,血脂,前列腺特异性抗原PSA水平及前列腺体积,骨量来评估疗效。乳腺癌与前列腺癌患者,血细胞比容>50%,未经治疗的严重的呼吸睡眠暂停综合征,严重的下尿道梗阻以及严重的心功能衰竭是睾酮替代治疗的禁忌证[45]。
对于年轻成年女性患者,可用雌孕激素序贯替代治疗,保持女性体态和月经来潮,最常用的替代疗法为口服雌二醇(2 mg/d)[46]。对于子宫结构完整的患者,还需要在每月初的10~12 d内加用甲孕酮10 mg/d,避免子宫内膜过度增生降低子宫癌变风险。对于年龄较大,不考虑月经来潮的女性患者,在完善宫颈刮片、乳腺超声和子宫卵巢超声后,如无其他禁忌证(高凝状态、乳腺癌家族史),可予以替勃龙1.25~2.5 mg/d口服。服药期间,应每年常规进行妇科检查。雌激素可降低皮质醇结合球蛋白数量,因此同时口服雌激素的女性患者应适当提高糖皮质激素剂量。
随访能及时发现肿瘤复发,对水电解质及内分泌状态进行及时的纠正和治疗。应在术后14、30 d,3、6个月及1年进行内分泌、电解质、肝肾功能及鞍区MRI检查(必要时增加随访频率),并且记录体质指数及生活质量评估结果。建议参考Duff等[33]或De Vile等[47]的生活质量评估量表,对神经系统功能、视力视野、垂体功能、下丘脑功能、精神心理,以及儿童受教育能力和成人工作能力等方面进行评估。1年以后,每年随访至少1次,除以上所有内容,还应包括骨龄(儿童)或骨密度(成人)检查。鉴于颅咽管瘤大部分在5年内复发,建议对所有患者随访至少5年。同时应注意患者的饮食摄入及体重情况,进行必要的相应控制,避免因下丘脑功能障碍,出现过度饮食,导致过度肥胖,出现相关并发症。
颅咽管瘤全切除后仍有一定的复发比例,次全切除、部分切除即使辅助放化疗后复发仍不可避免。肿瘤复发后,容易导致内分泌功能障碍、视力下降甚至失明。所以颅咽管瘤患者要及时复查,以避免或减少肿瘤复发导致的各种神经功能障碍。复发颅咽管瘤的生长方式与原发肿瘤的生长方式密切相关[30]。对于鞍内起源的Q型肿瘤,对蝶鞍内肿瘤包膜的不完全切除,是容易导致肿瘤复发的原因。对于结节漏斗部起源的T型肿瘤,为保护下丘脑结构,容易导致肿瘤残存,这部分患者也比较容易复发。复发颅咽管瘤可以在不加重内分泌障碍的基础上再次手术,建议再次全切除术治疗。建议选择有利于全切除肿瘤的入路进行手术,以更好地暴露肿瘤,并且避免在处理前一次手术造成的粘连而浪费过多的时间和精力。多次复发又难以全切除的患者可选择放射治疗。
本共识编写专家组成员名单(按姓氏拼音为序):蒋传路(哈尔滨医科大学附属第二医院神经外科)、窦长武(内蒙古医学院第一附属医院神经外科)、洪涛(南昌大学第一附属医院神经外科)、江涛(北京市神经外科研究所)、况建国(南昌大学第一附属医院神经外科)、兰青(苏州大学附属第二医院神经外科)、雷鹏(兰州军区总医院神经外科)、李维平(深圳大学第一附属医院神经外科)、林志雄(首都医科大学三博脑科医院神经外科)、刘云会(中国医科大学附属盛京医院神经外科)、刘志雄(中南大学湘雅医院神经外科)、马驰原(南京军区总医院神经外科)、马杰(上海新华医院小儿神经外科)、马文斌(中国医学科学院北京协和医学院北京协和医院神经外科)、马晓东(解放军总医院神经外科)、毛庆(四川大学华西医院神经外科)、毛颖(复旦大学附属华山医院神经外科)、牛朝诗(安徽省立医院神经外科)、潘军(南方医科大学南方医院神经外科)、潘力(复旦大学附属华山医院放射治疗科)、潘亚文(兰州大学附属第二医院神经外科)、漆松涛(南方医科大学南方医院神经外科)、邱炳辉(南方医科大学南方医院神经外科)、邱晓光(首都医科大学附属北京天坛医院放射治疗科)、石祥恩(首都医科大学三博脑科医院神经外科)、王伟民(广州军区总医院神经外科)、吴安华(中国医科大学附属第一医院神经外科)、伍学焱(中国医学科学院北京协和医学院北京协和医院内分泌科)、邢俭(解放军武警部队北京武警总医院小儿神经外科)、徐国政(解放军武汉总医院神经外科)、杨学军(天津医科大学总医院神经外科)、尤永平(江苏省人民医院神经外科)、张剑宁(解放军海军总医院神经外科)、张晓彪(复旦大学附属中山医院神经外科)、张亚卓(北京市神经外科研究所)、张玉琪(清华大学玉泉医院神经外科)、张志文(解放军第三○四医院神经外科)、赵刚(吉林大学附属第一医院神经外科)
本共识撰写者名单:漆松涛(南方医科大学南方医院神经外科)、伍学焱(中国医学科学院北京协和医学院北京协和医院内分泌科)、潘军(南方医科大学南方医院神经外科)、包贇(南方医科大学南方医院神经外科)、邱炳辉(南方医科大学南方医院神经外科)、彭俊祥(南方医科大学南方医院神经外科)、刘忆(南方医科大学南方医院神经外科)
























