
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
例1 先证者,男,38岁。因双眼视力逐渐下降28年于2013年2月1日来院就诊。患者自幼听力差,无渐进性听力下降病史,父母非近亲结婚,孕产史无特殊,幼时无发热、无使用特殊药物等病史。兄弟姐妹共9人,其中男性7人,女性2人,有一弟患有类似疾病,其余兄弟姐妹正常。除双侧中度耳聋外,全身检查无异常发现,男性生殖器发育正常。眼科检查:视力,右眼手动/30 cm,左眼手动/40 cm,光定位准确矫正视力不应。眼压,右眼17 mmHg,左眼15 mmHg。双眼外观正常,角膜透明,前房深度正常,房水清,瞳孔等大等圆,直径约4 mm大小,对光反射迟钝,双眼晶状体皮质呈灰白色混浊,玻璃体透明,眼底后极部青灰色,赤道部、周边部及后极部均可见大量骨细胞样色素沉着,视网膜动脉普遍变细,黄斑中心凹反光消失。视网膜电图(ERG)和闪光视觉诱发电位(F-VEP)检查双眼均为熄灭型。前庭功能检查无阳性发现。诊断:Usher综合征(Ⅱ型),双眼白内障。
例2 先证者弟,男,21岁。双眼进行性视力下降9年,自幼双耳聋,无渐进性加重,全身检查除双侧重度耳聋外无异常发现,男性生殖器发育正常。矫正视力,右眼-2.50 DS+0.50 DC×180°→0.1,左眼-2.00 DS→0.3。眼压,右眼11 mmHg,左眼11 mmHg。双眼外眼正常,角膜透明,前房深度正常,瞳孔直径3 mm,对光反射略迟钝,晶状体混浊(+),双眼底检查见赤道部、后极部骨细胞样色素沉着,视网膜动脉变细,黄斑中心凹反光消失。视野检查:双眼视野向心性缩小。诊断:Usher综合征(Ⅱ型),双眼白内障,双眼屈光不正。
例3 先证者姑母,女,59岁。双眼进行性视力下降46年,先天聋哑,父母非近亲结婚。全身检查除双侧重度耳聋外无异常发现。视力,右眼光感,左眼手动/20 cm,眼压,右眼18 mmHg,左眼18 mmHg。双眼外观正常,晶状体核性混浊,眼底模糊可见后极部骨细胞样色素沉着,视乳头色蜡黄,视网膜动脉极细,黄斑中心凹反光消失。视网膜电图(ERG)和闪光视觉诱发电位(F-VEP)检查双眼均为熄灭型。前庭功能检查无阳性发现。诊断:Usher综合征(Ⅱ型),双眼白内障。
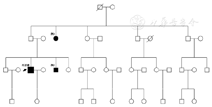
图例:■代表男性●代表女性
三例患者均转诊到耳鼻喉科验配助听器,建议例3患者白内障手术,与患者及家属沟通并告知因存在Usher综合征,白内障术后视力预后欠佳,患者拒绝接受手术治疗。考虑到三例患者是同一家系,建议接受基因检查,所有患者均拒绝检查。
Usher综合征(usher syndrome,USH)又称先天性聋视网膜色素变性综合征,是一种常染色体隐性遗传疾病。主要表现为不同程度的感音性神经性耳聋和视网膜色素变性(retinitis pigmentosa,RP),伴有或不伴有前庭功能障碍。文献报道的发病率约为1/25 000[1]~1/6 000[2],是遗传性聋哑的最常见原因。临床上,根据听力障碍的严重程度、是否存在前庭功能障碍和RP发病的年龄,分为Ⅰ型(USH1)、Ⅱ型(USH2)和Ⅲ型(USH3):Ⅰ型的主要特征是先天性重度感音神经性障碍、前庭反应消失和青春期前发作的RP;Ⅱ型特征是先天性中重度感音性神经性聋、前庭反应正常,青春期或之后发生双眼RP;Ⅲ型是进行性感音性神经性聋,前庭反应正常,RP发生的时间和程度表现各异[1]。Ⅰ型、Ⅱ型在临床上最常见,Ⅰ型占33%~44%,Ⅱ型占56%~67%[3]。Ⅱ型患者听力障碍通常出现在20岁以前,出现视觉症状的时间比Ⅰ型晚,夜盲出现的年龄是2~15岁,视网膜色素变性的诊断年龄平均为24岁[4]。
在遗传学上,Usher综合征一般为常染色体隐性遗传,视网膜的光感受器和内耳毛细胞都是有纤毛的神经表皮细胞,相关的基因位点有12个,有9个致病基因位点,其中5个USH1基因,MYO7A、USH1C、CDH23、PCDH15和USH1G,编码肌动蛋白相关的运动蛋白肌凝蛋白Ⅶa,3个USH2基因编码跨膜蛋白和G蛋白偶联受体-1(Vlgr1)和PDZ结构域包含蛋白whirlin;USH3A编码四次跨膜区域蛋白CLRN1[1,3,5,6,7]。USH基因编码的蛋白质组成一个USH蛋白质网络,USH蛋白质网络中相互作用的任何一个蛋白发生缺失或功能障碍,均可造成网络功能紊乱,导致内耳和神经视网膜感觉上皮变性,从而引起听觉和视觉的损害,导致Usher综合征的发生[1,3]。目前针对Usher综合征除了验配助听器和人工耳蜗植入外,没有其他有效的治疗方法。文献报道在动物模型上开展了一些实验性治疗,包括人工视网膜植入、药物治疗(人工合成的氨基糖苷类提前终止密码子124,PTC124)、细胞转化法或视网膜细胞移植、基因治疗,尚未取得突破性进展[1]。
本病例为一家系两代三人患病,双耳听力呈中重度感音性神经性聋,青春期前发生双眼视网膜色素变性,符合Usher综合征Ⅱ型诊断,均推荐患者转诊到耳鼻喉科验配助听器。遗憾的是,三例患者均拒绝接受基因检测,因此未能进一步从基因层面进行探讨。在临床上,早期诊断Usher综合征有助于发现家族性病例,便于进行遗传咨询和必要的助听器配戴,提高患者生活质量。

























