
Aarskog-Scott综合征是一种罕见病,典型表现为特殊面容、矮小、性腺发育不良及骨骼畸形等。本文报道1例男性患儿,初次就诊年龄为3岁6个月,主因"发现身高增长缓慢3年"就诊。围生期正常。智力运动发育大致正常。身长90 cm(<第3百分位,-3 SD),特殊面容,关节过伸,双手短粗,脚趾球形。披肩样阴囊,左侧隐睾。FGD1基因第9~12外显子存在1.1 Mb缺失变异,来源于母亲,符合Aarskog-Scott综合征的诊断。患儿自5岁起开始重组人生长激素治疗,治疗18个月,身高增长17 cm,无明显不良反应。当矮小患者同时伴有特殊面容、骨骼畸型及性腺发育不良三联征时,应考虑Aarskog-Scott综合征的可能。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Aarskog-Scott综合征(OMIM # 305400),也称为Aarskog综合征、面容-生殖器发育不良综合征(faciogenital dysplasia),是以特殊面容、手指及生殖器畸形为主要特征的一种临床综合征,于1970年被Aarskog[1]在挪威首次报道。目前为止,国内外基因确诊不足百例,每年新增基因确诊病例约为2~3例,无确切发病率统计,预估在1/25 000以下[2]。本研究分析1例本院诊断的Aarskog-Scott综合征诊疗经过,并进行文献复习。
患儿男性,年龄3岁6个月,第1胎第1产,足月顺产,出生体重2 600 g,出生身长49 cm。生后无窒息。主因"发现身高增长缓慢3年"入院。1岁时身高73 cm,2岁时身高80 cm,3岁身高87 cm。患儿3个月会抬头,6个月会坐,8个月会爬,1岁会走,会叫爸爸妈妈。2岁行倒睫手术。家族史:否认家族遗传病史。父母体健,无特殊面容。父亲身高182 cm,30岁,母亲身高164 cm,32岁,16岁月经初潮。
入院体格检查:体重13.5 kg(<第3百分位),身长90 cm(<第3百分位,-3 SD),体重指数16.6 kg/m2(第50~85百分位)。体型匀称,上部量50 cm,下部量40 cm,指尖距86 cm。特殊面容,圆脸,美人尖,眼距宽,上睑下垂,鼻翼宽扁,鼻孔前倾,长人中,下唇下端横向折痕,牙列间隙宽,高腭弓(图1A)。胸廓无畸形。双手短粗,指关节过伸;双脚扁宽,脚趾头呈球型(图1B、图1C),小腿粗壮。男童外生殖器,阴茎2 cm,披肩样阴囊,阴囊皱襞紧,左侧睾丸未触及,右侧睾丸容积1 ml。
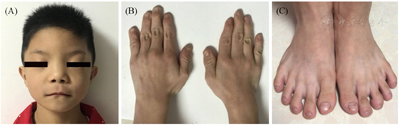
注:(A)面部特征,圆脸,眼距宽,上睑轻度下垂,鼻孔前倾,长人中,下唇下端横向折痕;(B)双手指短粗;(C)双脚趾呈球形
辅助检查:血常规、肝肾功能、电解质均正常。促甲状腺素1.79 IU/L(参考范围0.8~5 IU/L,下同)、游离T4 15.5 pmol/L(10.8~20 pmol/L)。促肾上腺皮质激素13.1 pg/ml(7.2~63.3 pg/ml)、皮质醇节律为8∶00时11.88 μg/ml(6.2~19.4 μg/ml)、生长激素激发试验峰值8.25 ng/ml(正常>10 ng/ml)。类胰岛素样生长因子1为54.2 ng/ml(49~289 ng/ml)、胰岛素生长因子结合蛋白3为2.78 μg/ml(0.9~4.3 μg/ml)。黄体生成素0.44 IU/L(<1.4 IU/L)、卵泡刺激素1.27 IU/L(<3.1 IU/L)、睾酮<0.087 nmol/L(<1.12 mmol/L)。人绒毛膜促性腺激素刺激试验后睾酮为14.9 nmol/L。曲普瑞林激发试验黄体生成素峰值<5 IU/L(符合青春期前水平)。染色体:46,XY。脊柱正侧位:未见脊柱侧弯。骨龄:4岁。垂体增强磁共振成像(MRI)及头颅MRI未见异常。睾丸B超:左侧腹股沟处可见12.6 mm×5.9 mm的异常回声,形态呈椭圆形,境界清楚,边缘光滑。右侧睾丸为14.2 mm×7.4 mm,位于阴囊内。心脏超声:未见异常。0~6岁小儿神经心理检查:发育商为90。结合患儿有特殊面容、骨骼发育异常、性腺发育不良等特点,疑诊矮小相关的临床综合征。暂未给予治疗,门诊随诊生长发育情况。全外显子测序:患儿FGD1基因第9~12外显子存在1.1 Mb纯和缺失变异。患儿之父样本在FGD1基因第9~12号外显子区域未见明确缺失变异,患儿之母样本在FGD1基因第9~12号外显子区域存在杂合缺失变异(图2)。符合Aarskog-Scott综合征的诊断。
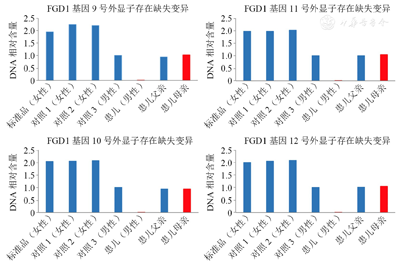
注:患儿FGD1基因9-12外显子脱氧核糖核酸(DNA)相对表达量低于其父及正常对照,其母表达量低于对照女性
4岁复诊时身高92 cm,4岁6个月复诊时身高95 cm,5岁复诊时身高99 cm,循既往生长曲线生长,显著低于遗传身高曲线。由于患儿上幼儿园有自卑心理,监测生长发育持续低于正常身高第3百分位以下,远低于遗传靶身高曲线,家属强烈要求应用生长激素,5岁起给予长效重组人生长激素治疗(起始剂量0.2 mg·kg-1·周-1)。应用生长激素治疗至今18个月,身高增长17 cm。目前6岁6个月,身高116 cm(图3)。应用生长激素期间无不良反应。上学后学习成绩中等。
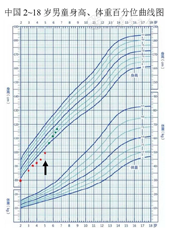
注:红点提示未治疗前身高增长情况,绿点提示治疗后身高增长情况;黑色箭头为开始应用生长激素治疗点
Aarskog-Scott综合征的遗传方式多样,主要为X染色体隐性遗传方式,也可为常染色体显性遗传及常染色体隐性遗传方式[1,2]。Aarskog[1]在1970年首次报道一种新的临床综合征,涉及1个家系中7例男性患者,均具有特殊面容及生殖器发育不良,1971年Scott[3]将这一疾病进一步完善。目前全球基因明确诊断患儿不足百例。
Aarskog-Scott综合征患者主要为男性,典型临床表现为特殊面容、骨骼畸形以及外生殖器发育异常三联征。目前临床诊断标准沿用1993年Teebi等[4]提出的标准,包括主要诊断标准和次要诊断标准。主要诊断标准:眼距过宽,短鼻子/鼻孔,上颌骨发育不良,下唇折痕,双手短粗,轻度指蹼,身材矮小,披肩样阴囊,第五手指短、弯曲。次要诊断标准:额头美人尖,上睑下垂,睑裂向下倾斜,耳位后悬,关节过伸,宽脚、脚趾球形,腹股沟隐睾症,脐疝。除诊断标准所描述的临床表现,患儿还可有尿道下裂、严重脊柱侧弯、牙齿早脱落及牙齿发育不良、先天性关节挛缩、先天性心脏病如法洛四联症、小头畸形、社会适应能力及语言发育落后,有些表现为兴奋、多动、注意力缺陷障碍[5,6,7,8,9,10]。生长发育迟缓是本病常见的就诊原因,生长激素刺激试验在Aarskog-Scott综合征患儿中多反应正常,患者往往表现为青春期发育延迟,终身高通常在-2 SD~-3 SD,但生育能力正常[11]。本病需要和Noonan综合征、Robin综合征、Optiz综合征、假性甲状旁腺功能减退症等疾病相鉴别,因为这些疾病和Aarskog-Scott综合征有重叠表现,诸如矮小、性腺发育不良、骨骼畸形等。仅通过临床诊断Aarskog-Scott综合征可能和实际发病情况有一定偏差。以上综合征在治疗和预后方面也与Aarskog-Scott综合征有很大差异,如Noonan综合征,肿瘤发生率高于普通人群,应用生长激素需更加谨慎,心血管系统受累概率高,故完善基因检测有利于治疗的选择以及个体化随访。
迄今为止,FGD1突变是唯一被证实导致Aarskog-Scott综合征的基因。FGD1位于Xp11.21,含18个外显子,961个氨基酸,编码Rho细胞分裂控制蛋白42(Cdc42)的特定鸟嘌呤核苷酸交换因子(GEF),与Rho的三磷酸鸟苷酶(gtp)及Cdc42特异性结合,刺激Cdc42异丙基化形式的gtp-gtp交换,对胚胎发生至关重要[12]。FGD1基因编码的蛋白质包含1个RhoGEF同源域、2个普列克底物蛋白同源域和1个FYVE锌指域[13]。Cdc42控制细胞骨架依赖的膜重排、转录激活、分泌膜转运、细胞周期中的G1转化和致肿瘤转化作用[14]。FGD1可以刺激有丝分裂原激活蛋白激酶级联反应,导致c-Jun末端激酶/应激活化蛋白激酶(JNK/SAPK)活化,主要在子宫内膜、脑组织、骨骼及其他25个组织表达[15]。FGD1蛋白在发育中的长骨的成骨活跃区域明显表达,基因表达缺失可能导致Aarskog-Scott综合征患者身材矮小[15]。而FGD1突变导致面部、生殖器发育不良以及骨骼畸形的细胞机制尚未完全清楚,但近期研究发现,FGD1/Cdc42信号通路参与细胞骨架和组织构成[15]。FGD1也是第一个被证实与注意力缺陷多动综合征相关的X连锁特异性分子缺陷[8]。FGD1突变类型包括错义突变、无义突变、小缺失插入、大片段缺失等。葛翼华等[16]报道首例中国基因确诊患儿,为年龄2岁2个月的男孩,因身材矮小伴特殊面容、单侧腹股沟疝气就诊,全外显子测序发现FGD1基因6号外显子c.1270 A>G错义突变,来源于母亲。本文患儿FGD1为涵盖9~12外显子1.1 Mb的缺失,为已经报道的致病性突变[13]。在首次分析数据时漏诊,提示对临床高度疑诊病例,当未发现点突变时,需考虑片段缺失的可能,进行线下分析数据,必要时采用多重连接探针扩增技术或实时荧光定量PCR以明确诊断。Orrico等[13]在涵盖110个疑诊本病的家系研究中,鉴定出24例发生致病突变,检出率为21.8%,其研究团队随后在46例临床诊断的患者中,检出9例患儿发生FGD1基因突变,检出率为19.56%[17]。以上2组涵盖病例最多的研究结果提示,该基因目前检出率在20%左右。由于本病和多种综合征有非常类似的临床表现,将其他综合征误诊为Aarskog-Scott综合征也是导致基因检出率低的一个原因[18,19]。此外,是否还有其他基因突变参与致病尚不明确。GEFs包含一系列调控蛋白家族,已在整个人类基因组中鉴定出21种,GEFs控制细胞内的各种过程,如基因表达、细胞骨架的重新排列、细胞内转运和代谢[20];这就可以解释本病临床的复杂多样性。迄今为止,未发现基因型和表型的相关性。
随着产前技术的不断发展,很多预后不良的遗传性疾病有望通过产前检查得以适时干预。然而,Aarskog-Scott综合征不同于其他严重或致死性遗传性疾病,有些患者临床表现轻微,在一个家系内相同基因变异导致的临床症状异质性很强,故针对本病的产前遗传咨询需非常慎重。目前还没有针对Aarskog-Scott综合征的对因治疗。与大多数临床综合征类似,治疗采用多学科合作对症方案。尿道下裂、腹股沟疝或脐疝、隐睾和严重的颅面部畸型可通过手术解决。如有颈椎神经根受压、第一颈椎发育不全、后弓不全、滑膜狭窄、齿状突畸形等,需和骨科医生商定解决方案。生长激素的治疗效果仅在初步研究中有所报道,对于生长发育和最终身高似乎具有积极影响[21]。然而,由于病例数量极少且缺乏随机对照的研究结果,需要更多患儿治疗数据以得出可靠结论。在神经发育方面,有些患儿可伴有轻度智力障碍和(或)注意力缺陷多动障碍(ADHD),需在神经科医生指导下给予干预。本病预后良好。其中,神经系统表现可能是需要长期随访关注的主要问题。
所有作者均声明不存在利益冲突

























