
报道3例以中枢性尿崩为首发表现的朗格汉斯细胞组织细胞增生症(LCH)患者,总结其临床表现、实验室检查、影像学检查、病理结果、诊断过程和治疗反应。3例患者早期均以中枢性尿崩症起病,垂体磁共振成像(MRI)均表现为垂体柄增粗,垂体后叶正常高信号消失。2例患者表现为孤立性下丘脑-垂体病变,1例表现为垂体和甲状腺多系统受累。病理结果显示典型的朗格汉斯细胞,免疫组织化学示S-100、CD1a、Langerin阳性。LCH临床表现呈现明显的异质性,容易误诊和漏诊。确诊依赖病理结果,孤立性下丘脑-垂体病变活检难度较大,建议积极筛查其他器官增加活检概率。LCH导致的神经垂体损害通常需要终生激素替代治疗。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
中枢性尿崩症(central diabetes insipidus)是下丘脑、垂体柄和垂体后叶病变造成抗利尿激素(antidiuretic hormone)合成、转运、储存及释放障碍而导致的临床症候群,主要表现为多尿、烦渴、多饮和低渗透压尿。抗利尿激素主要由下丘脑视上核、室旁核的大细胞神经元合成和分泌。室旁核神经元轴突横向汇入视上核发出的轴突,再向内侧下后方延伸,通过垂体柄终止于垂体后叶,形成了抗利尿激素的运输通路。垂体后叶贮存和释放抗利尿激素,释放受到血浆渗透压、循环血量和动脉血压的调节。根据发病机制的不同,中枢性尿崩症临床病因众多。
下丘脑视上核和室旁核受损时,核内存活神经元数量下降导致抗利尿激素合成减少,不足以维持机体需求时将出现中枢性尿崩症症状。视上-垂体束、室旁-垂体束或垂体柄受损时,将引起部分或完全的抗利尿激素运输受阻,导致部分性或完全性中枢性尿崩症。垂体柄损伤部位和神经元缺失程度相关,当垂体漏斗部及以上水平损伤时神经元缺失更严重[1]。垂体后叶受损,使抗利尿激素贮存和释放障碍,也会引起中枢性尿崩症。中枢性尿崩症的临床病因有原发性、继发性和遗传性3种,其中最常见的是继发性,包括颅脑外伤或手术后、下丘脑-垂体或鞍旁的肿瘤、感染性疾病(结核、梅毒、脑炎等)、浸润性疾病(结节病、Wegener肉芽肿等)、垂体炎等。
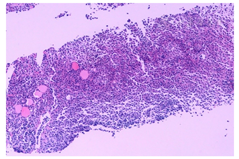
病例1:患儿男性,11岁5个月。2019年2月出现口干、多饮、多尿,饮水量达4 L/d,尿量与饮水量相当,夜尿2~3次,伴乏力、食欲下降、偏好冷饮水果,无头痛、视物模糊、视野缺损。入住本院后,查血常规、肝功能、肾功能、血电解质无异常,24 h尿量4.5 L,血渗透压327 mmol/L(参考范围280~310 mmol/L,下同)、尿渗透压129 mmol/L(40~1 200 mmol/L)、尿比重1.005(1.01~1.03)。禁饮试验过程中血钠水平升至164 mmol/L,终止试验。应用弥凝片治疗,症状明显改善。甲状腺功能:游离三碘甲状腺原氨酸(FT3)5.12 pmol/L(3.28~6.47 pmol/L)、游离甲状腺素(FT4)14.76 pmol/L(7.9~18.4 pmol/L)、促甲状腺激素(TSH)1.57 μIU/mL(0.34~5.6 μIU/mL)。甲状旁腺激素(PTH)28.24 pg/mL(15~65 pg/mL)。促肾上腺皮质激素(ACTH)水平8:00为44.3 pg/mL(7.0~61 pg/mL)、16:00为25.2 pg/mL。皮质醇水平:8:00为25.2 μg/dL(7~27 μg/dL)、16:00为17.4 μg/dL。催乳素29.9 ng/mL(3.5~19.4 ng/mL)、生长激素1.63 ng/mL(0.06~5 ng/mL)、胰岛素样生长因子1(IGF-1)水平160.7 ng/mL(117~329 ng/mL)。垂体磁共振成像(MRI):垂体柄上部局限性增粗,增强后明显强化,垂体后叶高信号未见显示,诊断为"中枢性尿崩症"(图1A)。甲状腺彩超:右侧叶中下部实性低回声结节,大小约25 mm×9 mm×10 mm,边界清,形态不规则;左侧叶中下级背侧可探及一大小约18.7 mm×9.8 mm×8.7 mm实性低回声结节,边界清,形态规则;双侧锁骨上窝均可及一实性结节,右侧约13 mm×6 mm,左侧约19.8 mm×13 mm,边界清,形态不规则。行甲状腺右侧叶结节细针穿刺活检术,病理诊断为朗格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis, LCH;图2),免疫组织化学示CD68(部分+)、S-100(+)、CD1a(+)、Langerin(+)、Ki-67(60%+),实时荧光PCR及DNA测序未发现BRAF基因V600E突变。彩超:肝脏和脾脏无异常。肺部计算机断层扫描(CT):未见异常。全身骨显像:各部位骨骼未见异常放射性浓聚或稀疏区。正电子发射计算机断层显像(PET-CT):甲状腺双叶低密度结节代谢活跃(标准摄取最大值:12.9),符合LCH;右颈Ⅳ区肿大淋巴结代谢稍活跃(标准摄取最大值:3.4),前纵隔片状软组织密度影代谢活跃(标准摄取最大值:10.2),考虑病变累及淋巴结及胸腺。
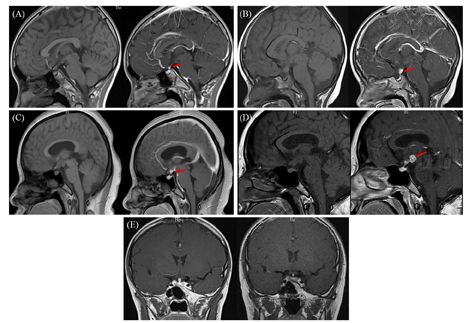
注:(A)病例1(初诊时),垂体柄上部局限性增粗,增强后明显强化,垂体后叶高信号未见显示;(B)病例2(初诊时),垂体柄增粗并结节状强化明显,垂体后叶高信号未见显示;(C)病例3(初诊时),垂体柄根部及第三脑室周围病变,增强后明显强化,垂体后叶高信号未见显示;(D)病例3(9个月后复诊时),垂体柄根部及第三脑室周围病变,范围较前扩大,垂体后叶高信号可见;(E)病例1治疗前、治疗1年后,垂体柄增粗较前缓解
病例2:患儿女性,6岁11个月。2岁3个月时出现烦渴、多饮、多尿,日饮水量约3 L,尿量约3 L,夜尿4~5次,伴饮食量下降(约1/3)和生长停滞。当地医院查头颅MRI:(1)垂体柄漏斗部增粗;(2)双侧颈部多发淋巴结肿大,诊断为"中枢性尿崩症"。口服弥凝片治疗,上述症状改善。2岁11个月时至本院查血常规、肝功能、肾功能、血电解质无异常。血渗透压301 mmol/L、尿渗透压166 mmol/L、尿比重1.005。甲状腺功能:FT34.70 pmol/L、FT410.84 pmol/L、TSH 2.36 μIU/mL。ACTH-皮质醇节律示ACTH水平8:00为12.1 pg/mL(7.2~63.3 pg/mL)、16:00为7.6 pg/mL,皮质醇水平8:00为481 ng/mL(171~536 ng/mL)、16:00为226 ng/mL。生长激素激发试验(左旋多巴):0 min为0.4 ng/mL、15 min为0.6 ng/mL、30 min为0.5 ng/mL、60 min为0.5 ng/mL、90 min为0.5 ng/mL。催乳素38.32 ng/mL(5.2~26.5 ng/mL);血β绒毛膜促性腺激素<0.1 mIU/mL(0~5 mIU/mL)、脑脊液β绒毛膜促性腺激素0.96 mIU/mL。复查垂体MRI(图1B):垂体柄增粗并结节状强化明显,大小约5.5 mm×5.8 mm×10.1 mm,考虑生殖细胞瘤、LCH、炎症;垂体后叶高信号未见显示。头颅X线片:未见明显异常。彩超:甲状腺未见异常;颈部多发肿大淋巴结、皮髓质分界清晰;肝脏和脾脏未见异常。行左侧颈部肿大淋巴结穿刺活检术,病理结果示较多成熟淋巴细胞、未见明确恶性病变。建议患儿家属进一步行垂体柄活检术,家属拒绝。5岁9个月时至外院查头颅MRI示鞍区结节样等信号影,增强后病变明显强化,大小约12 mm×8 mm×11 mm,行脑立体定位活组织检查术,病理示LCH,免疫组织化学示CD68(+)、S-100(+)、Langerin(+)、CD1a(少数+)、Ki-67(+)。
病例3:患者女性,30岁。2017年10月出现口渴、多饮、多尿,日饮水量5~6 L,尿量约5 L,夜尿3~4次,伴乏力、月经周期延长、月经量减少,无头痛、视物模糊、视野缺损,食欲正常,体重半年增加10 kg。患者20岁、22岁时分别行剖腹产分娩1子1女。入院后查血常规、肝功能、肾功能、血电解质无异常。血渗透压321 mmol/L(275~310 mmol/L)、尿渗透压150 mmol/L、尿比重1.005。甲状腺功能:FT34.88 pmol/L、FT46.25 pmol/L、TSH 0.47 μIU/mL。PTH:25.41 pg/mL。ACTH-皮质醇节律示ACTH水平8:00为30.5 pg/mL、16:00为28.0 pg/mL,皮质醇水平8:00为4.47 μg/dL、16:00为2.85 μg/dL,24 h尿(尿量5.2 L)游离皮质醇水平73 nmol/d(73~372 nmol/d)。生长激素<0.05 ng/mL,IGF-1水平199.1 ng/mL。性激素六项:卵泡刺激素(FSH)2.6 mIU/mL(3.03~8.08 mIU/mL)、促黄体生成素(LH)0.13 mIU/mL(2.39~6.60 mIU/mL)、雌二醇11.0 pg/mL(21~251 pg/mL)、孕酮<0.1 ng/mL(<0.1~0.3 ng/mL)、睾酮<0.13 ng/mL(0.11~0.57 ng/mL)。催乳素143.22 ng/mL;血β绒毛膜促性腺激素<0.1 mIU/mL。行禁饮加压素试验(+)。垂体MRI(图1C):垂体柄漏斗部及三脑室周围病变,增强后明显强化,考虑LCH、炎症性疾病、结节病;垂体后叶高信号未见显示。彩超:甲状腺未见异常,浅表淋巴结未见异常肿大,肝脏和脾脏无异常。肺部CT:未见异常。腰椎穿刺术失败,未能检测脑脊液β绒毛膜促性腺激素水平和病理细胞。建议患者行鞍区病变活检术,患者拒绝,给予糖皮质激素、左甲状腺素钠片(优甲乐)、醋酸去氨加压素片(弥凝片)替代治疗后出院。2018年6月至外院就诊,诊断为"垂体炎",予以甲泼尼龙(12 mg/d)口服,后调整为泼尼松(20 mg/d),症状无改善,渐出现继发性闭经。2019年1月至本院复查垂体MRI(图1D):垂体柄漏斗部及三脑室周围病变,范围较2018年4月扩大,垂体后叶高信号未见显示。2019年4月至外院行脑立体定位活组织检查术,病理示LCH,免疫组化S-100()、Langerin()、CD1a()、CD68(+)、催乳素(-)、Ki-67指数约10%。
LCH是以单核-巨噬细胞系统的朗格汉斯细胞(抗原提呈细胞)在1个或者多个器官增殖为特点的疾病,以脏器组织发生肉芽肿改变为临床特征。LCH多见于儿童,在儿童中的发病率为2~10/100万,成人中的发病率为1~2/100万,男性发病多于女性[2]。我国尚无该病的流行病学资料,本报道3例患者中,2例为儿童,2例为女性。LCH的发病机制尚未完全明确,关于树突状细胞是恶性转变还是炎症浸润仍有争论。目前发现的LCH相关基因包括BRAF-V600E、MAP2K1、BRAF-V600D、BRAF-600DLat、BRAF-T599A、MAP3K1等,上述基因突变均参与了LCH的发病机制[3]。不断发现的突变基因为恶性转变理论提供了更多证据,但不能解释部分LCH自限性的临床特征,因此炎症浸润理论仍不能忽视。
LCH可累及机体各个器官和系统,临床表现异质性大,典型症状者罕见。按病变范围分为两大类,累及单系统的LCH和累及多系统的LCH,单系统的LCH进一步分为单灶性病变和多灶性病变,多系统的LCH根据有无危险器官(肝脏、脾脏、血液系统,肺脏不再列为危险器官)受累分为高危组和低危组。本研究报道的3例患者中,病例1为多系统的LCH低危组,病例2为单灶性单系统的LCH,病例3为多灶性单系统的LCH。儿童LCH病变好发部位依次为骨骼(80%)、皮肤(33%)、垂体(25%)、肝脏(15%)、脾脏(15%)、造血系统(15%)、肺脏(15%)、淋巴结(5%~10%)以及不包括垂体的中枢神经系统(2%~4%)[4]。国内王涛等[5]的临床分析显示,122例LCH中,最常受累的器官为肺脏(54.92%),其他受累部位包括淋巴结(54.92%)、骨骼(36.07%)、皮肤(32.79%),垂体受累则少见(23.77%)。受累器官比例差异与研究对象相关,国内研究中成年患者多于未成年患者(89∶33)。
LCH相关骨骼病变主要是溶骨性破坏,也可能是成骨性损伤,各个部位骨骼均可能受累。颅骨为最常受累部位,其次是椎骨、肋骨、股骨。常见的临床表现为骨痛,其次为骨周软组织肿胀。X线一般表现为不均匀低密度灶,全身骨扫描可见异常浓聚或高放射灶。本研究报道的3例患者在发病初期筛查过程中,均未发现骨骼受累。任何年龄段均可出现肺部受累,成人较儿童多见,可能与成年患者吸烟相关[6]。临床表现为发热、干咳、活动后气促、呼吸困难等,影像学可显示为双肺弥漫性改变、肺间质纤维化、多发结节、多发囊性改变、多发蜂窝网格影、肺气肿等[5]。本文3例患者发病初期肺部CT筛查中肺部均未受累。
中枢神经系统中,LCH最常累及下丘脑-垂体区域,下丘脑因为血脑屏障作用较垂体少受累。下丘脑-垂体区域受累时最常见临床表现是中枢性尿崩症,中枢性尿崩症在LCH中发生率为5%~29.6%,其中不到5%的患者会同时发生神经垂体和腺垂体功能减退症[7,8]。本报道3例患者中,病例1单独出现中枢性尿崩症;病例2除了中枢性尿崩症外,还发生了生长激素缺乏症;病例3下丘脑和垂体均被累及,发生了中枢性尿崩症、继发性闭经、继发性甲状腺功能减退、继发性肾上腺皮质功能减退和成人生长激素缺乏症,提示病变范围和临床症状密切相关。MRI具有较高的空间分辨率和很好的组织分辨率,是目前显示下丘脑-垂体柄及其周围结构的首选方法,以矢状和冠状增强显示效果最佳。国内黄文献等[9]的研究显示,13例儿童孤立性垂体LCH的MRI中,垂体后叶T1W1正常高信号均消失,11例垂体柄呈结节样或均匀增粗,2例漏斗部呈结节样增粗,动态增强扫描呈渐进性强化、直至明显强化。本研究报道的3例患者MRI表现与上述研究类似。在缺乏病史的情况下,大多数LCH患者中枢神经系统的影像学改变是非特异的,需要和Rathke囊肿、鞍区肿瘤(脑膜瘤、错构瘤、生殖细胞瘤)、垂体微腺瘤等进行鉴别。
LCH经常伴发淋巴细胞性甲状腺炎,但罕见侵犯甲状腺,以弥漫性或结节性肿大为主,可伴发钙化,约70%伴有甲状腺功能减退[10,11]。本研究中病例1出现甲状腺受累,表现为结节性肿大,未见钙化灶,甲状腺功能正常。甲状腺LCH极少见,需要与淋巴细胞性甲状腺炎、甲状腺癌、慢性肉芽肿性甲状腺、淋巴瘤等进行鉴别,甲状腺细针穿刺活检联合免疫组织化学染色有助于鉴别诊断。也有研究报道,甲状腺LCH可与乳头状癌并存[12,13]。
LCH临床表现异质性大,缺少特异性,病变早期临床症状较轻,常被误诊或延误诊断。1987年国际组织细胞协会关于LCH的诊断标准为典型的组织病理及免疫病理学特征,即光镜下典型的组织形态学改变,免疫组化显示CD1a和S-100阳性。2013年欧洲指南推荐,以电子显微镜下发现细胞内的Birbeck颗粒作为诊断金标准[14]。近来研究发现,单克隆抗体langerin(CD207)可诱导Birbeck颗粒的形成,langerin(CD207)表达阳性可以代表Birbeck颗粒。当LCH累及骨骼、皮肤、肝脏、淋巴结等易于进行活检部位时,通过病理结果可以获得明确诊断。孤立性下丘脑-垂体受累的LCH因解剖位置的特殊性,活检难度和风险相对较高,限制了活检开展和普及,增加了诊断难度。因此,对于垂体柄受累的中枢性尿崩症患者,当MRI提示LCH可能性时,需积极筛查有无其他系统受累,增加活检概率和降低活检风险。
LCH的治疗应该结合临床表现和死亡风险综合考虑。确诊后首先进行危险分层以确定治疗的强度,根据病变部位和临床症状决定治疗方案。单灶性单系统的LCH通常只需要进行局部治疗,包括手术、放射、外用药物治疗。多系统LCH的治疗普遍以化学治疗为主,长春碱联合泼尼松龙是目前国际上的标准方案。标准方案无效的LCH患者,可尝试克拉屈滨、阿糖胞苷和氯法拉滨方案。随着LCH相关突变基因的发现,靶向治疗也成为一个新的治疗选择。本报道中3例患者在化学治疗和放射治疗后,尿崩症状均未获得改善,与既往研究结果一致[15,16];可能需要长期激素替代治疗。国内也有报道显示,垂体LCH患者在规律化疗后,停用醋酸去氨加压素片,尿量控制尚可[17]。
病例1患儿:2019年3月开始规律行阿糖胞苷联合克拉屈滨方案,化疗9次后改为静推长春地辛+口服巯嘌呤方案。治疗1年后复诊,患儿尿崩症状未缓解,身高增加5 cm,第二性征未发育。查FSH 0.48 mIU/mL、LH<0.10 mIU/mL、睾酮<0.025 ng/mL;生长激素2.45 ng/mL,IGF-1水平337.2 ng/mL。彩超:甲状腺双侧叶多发囊性小结节(TI-RADS:2级),双侧颈部未见异常肿大淋巴结。垂体MRI:垂体柄局限性增粗较前缓解,垂体后叶高信号未见显示。
病例2患儿:2019年2月开始行阿糖胞苷联合克拉屈滨方案,化疗8次后改为静推长春地辛+口服巯嘌呤方案,期间复查垂体MRI示鞍区病变明显改善,年身高增加14 cm。患儿现身高106 cm(低于同性别和年龄正常人群平均身高-2 SD),仍需醋酸去氨加压素片控制多饮、多尿症状,但剂量未进一步增加。
病例3患者:2019年5月至当地医院行鞍区放射治疗,放射剂量为20 Gy。放射治疗后,尿崩、继发性闭经、乏力症状未改善,仍需激素(醋酸去氨加压素片、醋酸泼尼松、左甲状腺素钠片、戊酸雌二醇片/雌二醇环丙孕酮片)替代治疗。2020年3月出现复视,当地医院查垂体MRI示病变侵犯视交叉,经本院血液科会诊后开始行环磷酰胺+阿霉素+长春新碱+醋酸泼尼松化疗方案。
临床初诊为中枢性尿崩症的患者,需常规行垂体MRI检查了解鞍区情况,对于下丘脑-垂体病变者应警惕LCH侵犯的可能性。LCH的确诊依赖病理学检查,下丘脑-垂体区域因为解剖位置的特殊性,活检难度较大,容易漏诊和误诊。临床诊断过程应积极筛查其他LCH可能累及但活检难度较低的器官,如骨骼、肺部、淋巴结、皮肤、甲状腺等,有利于早期诊断和治疗。LCH引起的下丘脑-垂体病变通常为不可逆损伤,需要长期激素替代治疗。
所有作者均声明不存在利益冲突

























