
先天性全身性脂肪营养不良1型(congenital generalized lipodystrophy type 1, CGL1)是由AGPAT2基因异常导致的常染色体隐性遗传疾病,主要临床表现为全身皮下脂肪消失、肌肉发达、皮下静脉明显、假性肢端肥大症、多毛、黑棘皮症等,常合并代谢性疾病,易被误诊为代谢综合征、2型糖尿病、多囊卵巢综合征、肢端肥大症及库欣综合征等。同时,与部分脂肪萎缩综合征的鉴别在临床上存在难度。本研究回顾分析1例CGL1患者的临床和遗传特征,并结合国内外文献进行分析总结,从而有助于加深对该罕见疾病的认识。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
先天性全身性脂肪营养不良(congenital generalized lipodystrophy, CGL)是一类罕见的常染色体隐性遗传代谢病,由Berardinelli[1]和Seip[2]最先报道,故又称Seip-Berardinelli综合征。据估计,CGL的人口患病率约为1/1 000万,多发生在父母近亲结婚的后代[3,4]。现已发现4种致病基因:AGPAT2、BSCL2、CAV1、PTRF,故目前临床上又分为4种亚型,分别为CGL1、CGL2、CGL3、CGL4[5]。其中,CGL1和CGL2最常见,约占95%[6]。CGL1系由位于染色体9q34的1-酰基甘油-3-磷酸O-酰基转移酶2(1-acylglycerol-3-phosphate O-acyltransferase 2, AGPAT2)基因突变引起[7]。AGPAT2具有催化溶血磷脂酸的酰化形成磷脂酸的作用,这是生物合成三酰甘油和甘油磷脂关键中间步骤[8,9,10]。因此,其功能异常可导致脂肪细胞三酰甘油生物合成和储存障碍、脂肪细胞凋亡加速,进而导致脂肪营养不良。本病最典型的临床特征为出生时或出生不久后全身代谢性脂肪组织如皮下、骨髓、胸腔及腹腔内脂肪组织几乎完全消失,而具有机械支持作用的脂肪组织如手心、足底、头皮、眼眶后关节周围的脂肪组织正常[11]。儿童期表现为食欲亢进、生长加速、甲状腺功能正常的高代谢状态[12]。常合并代谢紊乱,如糖尿病、高三酰甘油血症和肝脂肪变性等[13]。本研究介绍了1例AGPAT2复合杂合突变的CGL1患者,因其脂肪分布特征与典型的CGL1表现不尽相同,导致判断偏差,强调在对患者的临床表现进行评估时,不能仅通过对脂肪分布特征进行判断,必要时还须通过脂肪活检、基因检测等检测手段来辅助诊断,避免出现漏诊和误诊。
患者女性,26岁,未婚,糖尿病病程9年。2011年,患者以酮症起病,启动胰岛素降糖治疗,血糖控制不理想。2013年,患者因感染再次发生糖尿病酮症酸中毒,每日胰岛素使用加量,同时加用二甲双胍、格列美脲及罗格列酮口服药辅助降糖,血糖控制一般。该患者经检查发现三酰甘油水平明显升高,伴肝功能损伤,予"多烯磷脂酰胆碱"保肝,"非诺贝特"及"阿昔莫司"降血脂(具体剂量不详),经临床评估,不排除脂肪萎缩性糖尿病可能。2018年6月,患者出现眼部并发症,因眼底出血行眼底硅油填充术,胰岛素用量继续加大,调整为"门冬胰岛素(诺和锐)早餐前50 U、午餐前40 U、晚餐前50 U和重组甘精胰岛素(长秀霖)150 U qn",同时联合口服药物降糖。2019年2月患者因行卵巢囊肿手术术前准备,再次调整"重组甘精胰岛素180 U qn"。2020年6月,患者为系统评估其糖尿病病情再次入院,诊断"脂肪萎缩性糖尿病",同时评估糖尿病并发症,检查示糖尿病视网膜病变、糖尿病肾病、糖尿病周围神经病变。患者系足月顺产,出生无窒息抢救史,出生体重2 500 g,母乳喂养至6个月。患者1.5岁前经常腹泻、发热,2岁后体健。自幼臀部瘦、手脚细,皮肤黑,体毛多,生长发育较同龄人无异。12岁初潮,3~6个月1次,到18岁时1次/月,经量多。患者父母体健,非近亲结婚。家族中无类似疾病患者。
体格检查:身高161 cm,体重56.45 kg,体重指数21.78 kg/m2,体温36.4℃,血压134/79 mmHg (1 mmHg=0.133 kPa),消瘦貌、脸圆、发际低、头发浓密。皮肤较黑、多毛,双侧腋窝、腹股沟可见黑棘皮样改变,面部、颈部可见痤疮,全身见散在淡红色丘疹。心肺无异常,腹部膨隆,腹壁脂肪薄,肝脾肿大。四肢及臀部皮下脂肪明显菲薄(图1)。体脂仪示全身皮下脂肪减少(全身脂肪百分比为9.7%,内脏脂肪面积为59 cm2,腹部皮下脂肪面积为79 cm2)。
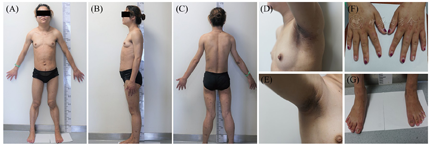
注:(A)全身正面;(B)全身侧面;(C)全身后面;(D)左侧腋窝;(E)右侧腋窝;(F)双手;(G)双足
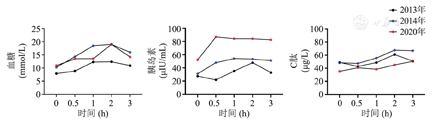
实验室检查(末次随访,2020年6月2日):肝酶升高[丙氨酸氨基转移酶23 IU/L(参考范围10~64 IU/L,下同)、天冬氨酸氨基转移酶30 IU/L(8~40 IU/L)、碱性磷酸酶33 IU/L(38~126 IU/L)、γ-谷氨酰基转移酶48 IU/L(7~64 IU/L)],血脂异常[三酰甘油16.01 mmol/L(0.56~1.70 mmol/L)、总胆固醇4.98 mmol/L(2.33~5.70 mmol/L)、高密度脂蛋白胆固醇0.52 mmol/L(0.80~1.80 mmol/L)、低密度脂蛋白胆固醇1.49 mmol/L(1.30~4.30 mmol/L)、载脂蛋白AI 0.98 g/L(1.06~1.88 g/L)、载脂蛋白B 0.89 g/L(0.46~1.13 g/L)、载脂蛋白E 8.6 mg/dL(2.9~5.3 mg/dl)]及尿常规异常[蛋白质(+)、24 h尿蛋白504 mg];HbA1C 8.6%(4.7%~6.4%)、口服葡萄糖耐量试验及同步血浆胰岛素、C肽释放试验提示严重胰岛素抵抗及糖耐量受损(图2);余未见异常。
影像学检查:腹部彩超显示脂肪肝、肝大(肝脏左叶长×厚为112 mm×103 mm,右叶斜径为131 mm);胸部CT显示右肺中叶结节、左肺内索条影、右侧水平裂增厚;盆腔彩超显示右卵巢已切除、左卵巢40 mm×31 mm×42 mm,内见无回声区单切面约14枚,直径3~13 mm;余未见明显异常。
基因检测结果:获得患者及家属知情同意后,提取患者外周血样送检全外显子基因组测序,并经一代测序验证患者及父母的突变位点。测序分析发现,患者AGPAT2基因2个等位基因复合杂合变异,等位基因c.356_362del:p.Ala119fs来源于父亲,目前尚无文献报道,参照美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics, ACMG)相关指南[14],评分为致病突变位点;等位基因c.683T>C:p.L228P来源于母亲,为已知的可能致病的变异位点[7,15]。患者家系图及基因测序结果见图3。
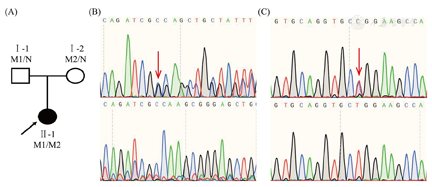
注:(A)家系图,黑色箭头所指为患者;(B)患者AGPAT2基因测序结果1(M1突变),红色箭头指示cDNA的第356至362位碱基缺失;(C)患者AGPAT2基因测序结果2(M2突变),红色箭头指示c.683位点的胸腺嘧啶(T)被胞嘧啶(C)取代;M1: c.356_362del:p.Ala119fs; M2:c.683T>C:p.L228P; N:正常
诊治结果:患者入院后予维格列汀+二甲双胍以提高胰岛素敏感性,促进外周组织利用葡萄糖,阿卡波糖抑制胃肠道碳水化合物的吸收,胰岛素逐渐减量为门冬胰岛素早餐前15 U、午餐前15 U、晚餐前10 U和重组甘精胰岛素60 U qn,联合钠-葡萄糖协同转运蛋白2(SGLT2)抑制剂恩格列净尿路排糖,患者空腹血糖水平稳定在3.8~5.9 mmol/L,同时予多烯磷脂酰胆碱保肝,非诺贝特、依折麦布、吉非罗齐、奥司利他随餐服用,以期降低三酰甘油水平。
本研究报道的CGL1患者脂肪萎缩以四肢为主,面颈部脂肪保留,与典型CGL1患者脂肪组织消失的模式不尽相同,同时与部分脂肪萎缩综合征临床表型有共同点,从而导致明确诊断的时间延迟;以"脂肪营养不良""lipodystrophy""AGPAT2""CGL1"为关键词在万方、中国知网、PubMed数据库查阅复合杂合突变的病例报道,共搜索到8篇文献,中文1篇、外文7篇,共16例,以期汇总CGL1复合杂合患者的临床特征的异同点(表1)[7,16,17,18,19,20,21,22]。其中,中国目前报道了2例复合杂合突变,本研究为第3例,本例与国内报道的另外2例的表型异同点:(1)相同为均有高三酰甘油血症、四肢肌肉发达;(2)差异为2019年报道的患儿以生长发育迟缓起病,暂未发生糖尿病,未用胰岛素治疗;2020年报道的患儿以多饮、多尿伴体重下降起病,糖尿病经胰岛素治疗,此2例脂肪萎缩为全身性,均有肌肉肥大、假性肢端肥大症、黑棘皮样改变等体征,而本例患者以糖尿病酮症起病,胰岛素治疗,后续胰岛素用量加大而血糖控制不佳,该患者脂肪萎缩以四肢为主,面部及躯干脂肪仍可见,说明不同变异类型对皮下脂肪分布的影响仍然未知。
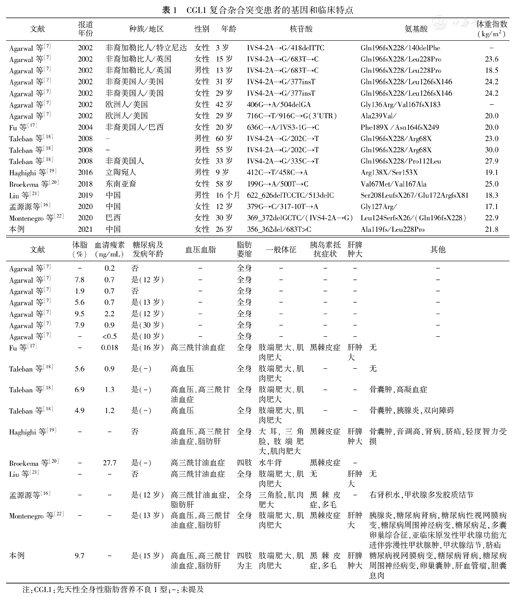
CGL1复合杂合突变患者的基因和临床特点
CGL1复合杂合突变患者的基因和临床特点
| 文献 | 报道年份 | 种族/地区 | 性别 | 年龄 | 核苷酸 | 氨基酸 | 体重指数(kg/m2) |
|---|---|---|---|---|---|---|---|
| Agarwal等[7] | 2002 | 非裔加勒比人/特立尼达 | 女性 | 3岁 | IVS4-2A→G/418delTTC | Gln196fsX228/140delPhe | - |
| Agarwal等[7] | 2002 | 非裔加勒比人/英国 | 女性 | 15岁 | IVS4-2A→G/683T→C | Gln196fsX228/Leu228Pro | 23.6 |
| Agarwal等[7] | 2002 | 非裔加勒比人/英国 | 男性 | 13岁 | IVS4-2A→G/683T→C | Gln196fsX228/Leu228Pro | 18.5 |
| Agarwal等[7] | 2002 | 非裔美国人/美国 | 女性 | 31岁 | IVS4-2A→G/377insT | Gln196fsX228/Leu126fsX146 | 24.2 |
| Agarwal等[7] | 2002 | 非裔美国人/美国 | 女性 | 29岁 | IVS4-2A→G/377insT | Gln196fsX228/Leu126fsX146 | 24.2 |
| Agarwal等[7] | 2002 | 欧洲人/美国 | 女性 | 42岁 | 406G→A/504delGA | Gly136Arg/Val167fsX183 | - |
| Agarwal等[7] | 2002 | 欧洲人/美国 | 女性 | 29岁 | 716C→T/916C→G(3′UTR) | Ala239Val/ | 20.0 |
| Fu等[17] | 2004 | 非裔美国人/巴西 | 女性 | 20岁 | 636C→A/IVS3-1G→C | Phe189X /Asn164fsX249 | 20.0 |
| Taleban等[18] | 2008 | - | 男性 | 60岁 | IVS4-2A→G/202C→T | Gln196fsX228/Arg68X | 23.0 |
| Taleban等[18] | 2008 | - | 男性 | 55岁 | IVS4-2A→G/202C→T | Gln196fsX228/Arg68X | 30.0 |
| Taleban等[18] | 2008 | 非裔美国人 | 女性 | 33岁 | IVS4-2A→G/335C→T | Gln196fsX228/Pro112Leu | 27.9 |
| Haghighi等[19] | 2016 | 立陶宛人 | 男性 | 9岁 | 412C→T/458C→A | Arg138X/Ser153X | 19.1 |
| Broekema等[20] | 2018 | 东南亚裔 | 女性 | 58岁 | 199G→A/500T→C | Val67Met/Val167Ala | 25.0 |
| Liu等[21] | 2019 | 中国 | 男性 | 16个月 | 622_626delTCCTC/513delC | Ser208LeufsX267/Glu172ArgfsX81 | 18.3 |
| 孟源源等[16] | 2020 | 中国 | 女性 | 12岁 | 379G→C/317-10T→A | Gly127Arg/ | 17.1 |
| Montenegro等[22] | 2020 | 巴西 | 女性 | 30岁 | 369_372delGCTC/(IVS4-2A→G) | Leu124SerfsX26/(Gln196fsX228) | 22.9 |
| 本例 | 2021 | 中国 | 女性 | 26岁 | 356_362del/683T>C | Ala119fs/Leu228Pro | 21.8 |
| 文献 | 体脂(%) | 血清瘦素(ng/mL) | 糖尿病及发病年龄 | 血压血脂 | 脂肪萎缩 | 一般体征 | 胰岛素抵抗症状 | 肝脾肿大 | 其他 |
|---|---|---|---|---|---|---|---|---|---|
| Agarwal等[7] | - | 0.2 | 否 | - | 全身 | - | - | - | - |
| Agarwal等[7] | 7.8 | 0.7 | 是(12岁) | - | 全身 | - | - | - | - |
| Agarwal等[7] | 1.9 | 0.7 | 否 | - | 全身 | - | - | - | - |
| Agarwal等[7] | 5.6 | 0.7 | 是(13岁) | - | 全身 | - | - | - | - |
| Agarwal等[7] | 9.5 | 2.2 | 是(12岁) | - | 全身 | - | - | - | - |
| Agarwal等[7] | 7.9 | 0.9 | 是(30岁) | - | 全身 | - | - | - | - |
| Agarwal等[7] | - | <0.5 | 是(10岁) | - | 全身 | - | - | - | - |
| Fu等[17] | - | 0.018 | 是(16岁) | 高三酰甘油血症 | 全身 | 肢端肥大,肌肉肥大 | 黑棘皮症 | 肝肿大 | 无 |
| Taleban等[18] | 5.6 | 0.9 | 是(-) | 高血压 | 全身 | 肢端肥大,肌肉肥大 | - | - | 无 |
| Taleban等[18] | 6.9 | 1.3 | 是(-) | 高血压,高三酰甘油血症 | 全身 | 肢端肥大,肌肉肥大 | - | - | 骨囊肿,高凝血症 |
| Taleban等[18] | 4.9 | 1.2 | 是(-) | 高血压 | 全身 | 肢端肥大,肌肉肥大 | - | - | 骨囊肿,胰腺炎,双向障碍 |
| Haghighi等[19] | - | - | 否 | 高血压,高三酰甘油血症,脂肪肝 | 全身 | 大耳,三角脸,肢端肥大,肌肉肥大 | 黑棘皮症 | 肝脾肿大 | 骨囊肿,音调高,肾病,脐疝,轻度智力受损 |
| Broekema等[20] | - | 27.7 | 是(-) | 高三酰甘油血症 | 四肢 | 水牛背 | 黑棘皮症 | - | |
| Liu等[21] | - | - | 否 | 高三酰甘油血症 | 全身 | 肢端肥大,肌肉肥大 | 无 | 肝肿大 | 无 |
| 孟源源等[16] | - | - | 是(12岁) | 高三酰甘油血症,脂肪肝 | 全身 | 三角脸,肌肉肥大 | 黑棘皮症,多毛 | - | 右肾积水,甲状腺多发胶质结节 |
| Montenegro等[22] | - | - | 是(13岁) | 高血压,高三酰甘油血症,脂肪肝 | 全身 | 肢端肥大,肌肉肥大 | 黑棘皮症 | 肝肿大 | 胰腺炎,糖尿病肾病,糖尿病性视网膜病变,糖尿病周围神经病变,糖尿病足,多囊卵巢综合征,亚临床原发性甲状腺功能亢进伴弥漫性甲状腺肿,甲状腺结节,脐疝 |
| 本例 | 9.7 | - | 是(15岁) | 高血压,高三酰甘油血症,脂肪肝 | 四肢为主 | 肢端肥大,肌肉肥大 | 黑棘皮症,多毛 | 肝脾肿大 | 糖尿病视网膜病变,糖尿病肾病,糖尿病周围神经病变,卵巢囊肿,肝血管瘤,胆囊息肉 |
注:CGL1:先天性全身性脂肪营养不良1型;-:未提及
本例患者最后经基因检测确诊为CGL1,父母均发现突变。患者是先天性AGPAT2基因的复合杂合突变,c.356_362del(p.Ala119fs)突变来源于父亲,为cDNA的第356至362位碱基缺失,终止密码子提前产生,导致蛋白质截短,或可能导致无义变异介导的mRNA降解,目前尚无文献报道;来源于母亲的c.683T>C(p.L228P)突变是错义突变,为cDNA的第683位碱基T替换为C,导致第228位密码子由编码亮氨酸变为编码脯氨酸,已有文献报道该突变[7,15]。本例与国内既往报道2例病例的表型存在异同点:(1)相同为均有肌肉肥大、高胰岛素血症、糖尿病、高脂血症的临床表现;(2)差异为本例与既往报道中1例均出现糖尿病、大剂量胰岛素治疗效果控制不佳、并发症严重,而既往报道的另1例发病年龄晚,且糖尿病发生后,仅经口服药物治疗后,未出现并发症,说明该基因突变的临床特征存在异质性,对该疾病的诊断需要对临床资料及基因资料进行整合分析。
AGPAT2由278个氨基酸组成,含2个高度保守的结构域NHXXXXD和EGTR,属酰基转移酶家族成员,存在于内质网上,在脂肪组织中高表达[7,23,24]。AGPAT2突变导致脂肪细胞三酰甘油合成和储存障碍,进而导致脂肪萎缩。此外,AGPAT2突变可影响与脂肪分化高度相关的过氧化物酶体增殖物激活受体(PPAR)-γ通路的信号转导,由此导致脂肪分化成熟障碍,引起皮下脂肪萎缩;同时影响磷酯酰肌醇3激酶(PI3K)/蛋白激酶B(Akt)通路,导致葡萄糖氧化障碍,引起严重的胰岛素抵抗[25]。
对于本病的诊断若只关注局部某一特点,而不对患者情况进行整合分析及对疾病追根溯源,则容易误诊为2型糖尿病、代谢综合征、严重胰岛素抵抗综合征、部分性脂肪萎缩等。对于糖尿病合并其他异常特征,如脂肪分布异常、血脂代谢紊乱、高胰岛素血症等,临床上应充分考虑特殊类型糖尿病,基因检测可发挥鉴别、诊断及分型的重要作用,通过发现异常基因突变位点确定疾病类型,指导临床治疗。本研究报道的患者,尽管改善胰岛素敏感性等对症治疗是可行的,但是若不经基因检测明确诊断,将无法对患者进行全面正确的判断和评估。通过基因检测发现患者AGPAT2基因的复合杂合突变:c.356_362del(p.Ala119fs);c.683T>C(p.L228P),其父母各出现其中一个突变。此发现有助于对患者的预后进行评估,也有助于评估该家系中其他成员患病情况。
目前,针对CGL1无特效治疗,仅对症治疗糖尿病、高脂血症等代谢异常。改善胰岛素敏感性的首要策略是调整生活方式,包括适当饮食控制和按需锻炼。除此之外,对于有脂肪营养不良伴糖尿病的患者,可应用二甲双胍和噻唑烷二酮类药物减轻高血糖和高脂血症[26,27,28,29]。然而,很多患者无法仅仅依赖口服药物实现良好的血糖控制,仍需要剂量很高的胰岛素治疗,常需使用浓缩型胰岛素。除此之外,患者需要控制高三酰甘油血症以降低胰腺炎风险,可单用贝特类药物或与他汀类药物联用改善。有研究显示,瘦素的替代治疗可以改善外周葡萄糖利用,并降低肝脏葡萄糖输出和肝脂肪变性,长期使用也可改善胰岛素抵抗[30,31]。2014年,美国食品药品监督管理局(FDA)批准美曲普汀(人瘦素类似物)用于治疗CGL或获得性全身性脂肪营养不良患者[32]。整形美容手术包括面部重建术(植入填充物"替代"脂肪组织)及抽脂和脂肪切除术(去除多余脂肪),可改善局部外观,提高患者生活质量,但是这些治疗方法效果各异且适应证尚不明确。
综上所述,CGL1是以全身代谢性脂肪缺失伴代谢紊乱为特征的AGPAT2基因突变导致的常染色体隐性遗传病。由于其机械性脂肪组织并未受累,临床医师若局限于患者局部表现,极易导致CGL1被误诊为部分性脂肪萎缩或漏诊。详细的体格检查和实验室检查有助于临床医师发现脂肪分布差异和代谢紊乱情况,从而进一步考虑特殊类型糖尿病,基因检测是此类疾病诊断的金标准,对此疾病实现早期诊断,采取合理营养、控制血糖及血脂、积极防治并发症等措施有助于改善预后。
所有作者均声明不存在利益冲突

























