
患者男性,39岁。因"腰痛8个月"入院。8个月前患者出现腰痛,伴乏力、肩颈痛,偶有心悸,无头痛、头昏,无四肢疼痛,至本院门诊就诊,测血钙2.69~2.78 mmol/L(正常值2.15~2.50 mmol/L,下同),门诊以"高钙血症"收住入院。自起病以来,患者精神、睡眠、饮食欠佳,大小便正常,体力下降、体重下降约5 kg。既往史:否认特殊病史,否认家族遗传病史,其父有高钙血症病史,无任何症状。体格检查:体温36.5℃,脉搏128次/min,呼吸20次/min,血压120/92 mmHg(1 mmHg=0.133 kPa),神志清楚,精神可,无关节畸形等,查体未见明显特殊。
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患者男性,39岁。因"腰痛8个月"入院。8个月前患者出现腰痛,伴乏力、肩颈痛,偶有心悸,无头痛、头昏,无四肢疼痛,至本院门诊就诊,测血钙2.69~2.78 mmol/L(正常值2.15~2.50 mmol/L,下同),门诊以"高钙血症"收住入院。自起病以来,患者精神、睡眠、饮食欠佳,大小便正常,体力下降、体重下降约5 kg。既往史:否认特殊病史,否认家族遗传病史,其父有高钙血症病史,无任何症状。体格检查:体温36.5℃,脉搏128次/min,呼吸20次/min,血压120/92 mmHg(1 mmHg=0.133 kPa),神志清楚,精神可,无关节畸形等,查体未见明显特殊。
入院检查:血钙2.69 mmol/L(2.15~2.55),血磷0.84 mmol/L(0.81~1.45),甲状旁腺素(PTH)49.32 pg/mL(15~65),低密度脂蛋白胆固醇3.55 mmol/L(<3.37),25-羟维生素D(含D2和D3) 14.9 ng/mL(14~20),总Ⅰ型前胶原氨基端肽77.9~91.0 ng/mL(9.06~76.24),I型胶原交联羧基端肽0.85 ng/mL(0.043~0.783)。24 h尿钙5.98 mmol(2.5~7.5),24 h尿磷12.8 mmol(12.9~42);性激素全套正常,肿瘤血清标记物均阴性,血清皮质醇节律正常。甲状旁腺显像(双时相法):未见典型甲状旁腺增生或腺瘤征象。甲状腺B超提示甲状腺实质回声不均。双能X线吸收测量法(DXA)骨密度显示腰椎Z-3.5,髋部Z值-1.7。腹部B超提示脂肪肝,左侧肾囊肿伴囊壁钙化。经过高通量测序筛选,滤除UCSC数据库Common SNPs,滤除dbSNP数据库及1000-genome-project数据库的常见突变,滤除无功能的同义突变和内含子突变,得到候选致病突变。对候选致病突变进行突变位点功能预测,保守性评估,Clinvar、HGMD等数据库检索,并经Sanger测序验证,患者及其父亲存在钙敏感受体(calcium-sensing receptor,CaSR)基因c.343G>A杂合突变,导致其编码氨基酸发生p.Val115Ile错义突变,经过数据库查询,该突变为CaSR基因新突变。患者父亲为携带该基因的杂合突变型,其母亲为野生型(图1)。诊断考虑:(1)家族性低尿钙高钙血症;(2)骨质疏松(维生素D不足)。
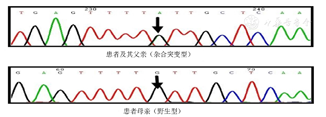
家族性低尿钙高钙血症(familial hypocalciuric hypercalcemia,FHH)是一类相对良性的疾病。临床表现为高血钙、低尿钙,往往呈家族聚集发病。虽然FHH是一种良性病变,但是由于临床症状并不特异,往往与原发性甲状旁腺功能亢进症(primary hyperparathyroidism,PHPT)症状重叠,容易漏诊和误诊。
FHH这一罕见疾病的病因是由于CaSR基因失活性突变。CaSR的某些残基突变为失活性并导致FHH,或为激活性并导致常染色体显性低钙血症,这取决于突变后的具体氨基酸,这种现象被称为开关突变[1]。FHH是由于CaSR基因失活性(或功能丧失性)突变,使钙-甲状腺旁腺曲线右移,此时需要较高的钙浓度才能抑制PTH的释放,导致血清钙浓度"重新设置"为高于正常的水平,所以引起高钙血症。
CaSR在很多组织中表达,包括甲状旁腺、肾脏、骨髓、破骨细胞和成骨细胞、乳腺、甲状腺C细胞、胃的胃泌素分泌细胞、肠、脑的部分区域以及其他组织细胞[2]。CaSR的主要功能之一是调节钙平衡,CaSR可感受血清钙离子浓度的细微变化,从而通过改变甲状旁腺及肾脏的功能来使血清钙浓度恢复正常水平[3]。
FHH受累的杂合子患者一般在儿童期发病[4],临床表现为偶然发现的轻度高血钙、低尿钙、PTH水平正常、血清镁水平正常或轻度升高。本例是中年男性,FHH(杂合基因突变)合并骨质疏松,进行了继发性骨质疏松相关检查。有研究报道在FHH中合并骨质疏松病史的比例高达8%,但是具体机制不太清楚[5]。
同样的,FHH临床表现并不特异。据报道,只在10%的FHH患者中表现高钙血症[6]。据报道1例54岁马来西亚女性[7],临床无特殊症状,仅体检表现偶然轻微高钙血症,低PTH,间断低血磷,低或者正常尿钙。患者的2个兄弟姐妹,1个侄儿子和儿子均为高钙血症,但不伴肾结石。该病例基因检测提示FHH1型。
FHH发病为家族聚集性,多见于儿童期发病,分为3型,其中FHH1型是最常见的类型,主要由于CaSR基因失活性突变。2012年报道就有150种失活性突变[8]。FHH3型和FHH2型都比较少见,分别由于AP2S1和GNA11基因突变引起[9]。有报道FHH3型出现在2个家族30岁以上的成员中。Vargas-Poussou等[10]分析了220例FHH患者,其中60%患者携带CaSR基因突变,9%患者携带AP2S1基因突变,31%患者没有发现GNA11基因突变。比较FHH1型和FHH3型临床特点,FHH3型肾小管尿钙重吸收率增高,有较高的总血钙水平,但是2种类型的血PTH、尿钙排泄率相类似。比较有趣的是,FHH3型患者身高比FHH1型偏矮。同时该团队发现,2种类型的PTH和25-羟维生素D,以及血清磷水平两者之间没有相关性。比较PHPT和FHH的临床特点,PHPT发病年龄较FHH偏大,FHH较PHPT血钙高、PTH低、尿钙排泄率低。因此,无论是FHH1型还是3型,FHH和PHPT之间的鉴别都有一定难度,血钙、血磷、PTH和尿Ca/Cr指标没有明确的界限。FHH占高血钙原因的1%~2%,其他原因中绝大多数都是PHPT,2种疾病的临床表现有所重叠。Eldeiry等[11]报道2例FHH合并PHPT患者,其临床表现为高血钙、低血磷、高PTH、低尿钙,虽然甲状旁腺手术切除不能解决高血钙,但是能缓解高钙血症症状以及由于甲状旁腺亢进所导致的潜在并发症。
Bertocchio等[5]采用回顾性研究建立了鉴别FHH和PHPT的风险模型,所有研究对象均为高钙血症,但PTH在正常范围之内。他们建立的风险模型综合血钙、PTH、血清骨钙素、24 h尿钙/肌酐清除率(24 h CCCR)。该风险模型用于鉴别FHH和PHPT明显优于24 h CCCR,其受试者工作特征曲线下面积(area under the receive operating characteristic curve, AUROC)为0.928时,对PHPT的诊断率高达100%,临床实践证实从85名该人群中排除PHPT(51名)后的34例患者行基因检测,均证实为FHH。也有学者建议根据24 h CCCR来鉴别,如果低24 h CCCR(1%)则考虑FHH,如果高24 h CCCR(2%)考虑PHPT[12]。但是Bertocchio等[5]认为24 h CCCR鉴别两者是不可靠的。FHH中2%的患者24 h CCCR值超过2%,只有26%的患者24 h CCCR小于2%。
那么,有没有其他的尿钙指标可以参考呢?英国学者[13]做了相关研究,纳入明确诊断的FHH(基因)和PHPT患者(手术病理),PHPT组24 h尿钙排泄(24 h UCE)平均值为8.3(5.6~11.2) mmol/24 h,FHH 3.2(2.1~6.1) mmol/24 h(P<0.001)。PHPT组24 h CCCR为0.02(0.013~0.026),FHH组为0.01(0.002~0.02,P=0.001)。24 h UCE(切点≥2.5 mmol/24 h)对PHPT的灵敏度是96%,24 h CCCR(切点>0.02)为47%。24 h UCE(切点<2.5 mmol/24 h)对FHH的特异度为29.4%,24 h CCCR(切点<0.02)为93%。因此,该研究认为24 h UCE对诊断PHPT更为敏感,但是24 h CCCR诊断FHH更为特异度。可见,诊断FHH和PHPT时应结合多种指标综合分析(表1)。
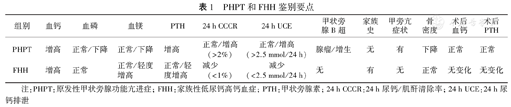
PHPT和FHH鉴别要点
PHPT和FHH鉴别要点
| 组别 | 血钙 | 血磷 | 血镁 | PTH | 24 h CCCR | 24 h UCE | 甲状旁腺B超 | 家族史 | 甲旁亢症状 | 骨密度 | 术后血钙 | 术后PTH |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PHPT | 增高 | 正常/下降 | 正常/下降 | 增高 | 正常/增高(>2%) | 正常/增高(>2.5 mmol/24 h) | 腺瘤/增生 | 无 | 有 | 下降 | 正常 | 正常 |
| FHH | 增高 | 正常 | 正常/轻度增高 | 正常/轻度增高 | 减少(<1%) | 减少(<2.5 mmol/24 h) | 无 | 有 | 无 | 正常 | 无变化 | 无变化 |
注:PHPT:原发性甲状旁腺功能亢进症;FHH:家族性低尿钙高钙血症;PTH:甲状旁腺素;24 h CCCR:24 h尿钙/肌酐清除率;24 h UCE:24 h尿钙排泄
FHH合并PHPT患者是否应该行甲旁亢手术?德国学者Frank-Raue等[14]对139例高钙血症患者行基因检测发现其中135例是FHH,另外4例是FHH合并PHPT。这4例患者行甲旁亢手术后25%患者血钙下降,75%患者恢复到正常。FHH合并PHPT的共存机制并不太清楚,可能因为CaSR的失活性突变导致CaSR数量的改变,而该CaSR与甲状旁腺细胞增殖相关。美国学者Majumdar等[15]报道一对母女,先证者(女儿)表现乏力、骨痛,生化提示轻度的高钙血症,较低尿钙排泄率,骨化三醇增高,低或正常的PTH,先证者(女儿)诊断囊性纤维性骨炎和结节病,但是女儿经过甲状旁腺手术后发现甲状旁腺正常,虽然高血钙和骨化三醇指标没有改善,但是患者临床症状有所缓解。母亲没有任何临床症状,行基因检测提示CaSR新的突变。但是以上病例数仍然偏少或是个例,FHH合并PHPT患者是否应该行甲旁亢手术还需要大样本研究探讨。
FHH治疗的主要目的是为改善症状,对症处理。但是严重高钙血症难以缓解,还是需要药物干预治疗。钙代谢紊乱在老年人中常见。手术对于FHH并不是好的选择,但是拟钙剂的适应证主要是成年FHH患者,也有个案报道在婴幼儿中由于常规降钙处理无效,而使用拟钙剂使血钙水平降低恢复到正常水平[16]。FHH的自然病程通常呈良性,且甲状旁腺次全切除不能治愈FHH,因此大多数患者不应进行颈部探查和其他干预措施。临床医师应当对受累的FHH家族成员进行随访,解释该病的良性性质,避免不必要的甲状旁腺手术。
所有作者均声明不存在利益冲突

























