
McCune-Albright综合征(McCune-Albright syndrome, MAS)是一种非常罕见的疾病,其典型临床特点是骨纤维异样增殖症(fibrous dysplasia, FD)、皮肤牛奶咖啡斑和(或)内分泌器官功能紊乱三联征。近期研究表明,MAS的主要发病机制是鸟苷酸结合蛋白的刺激型α亚基(Guanine nucleotide-binding protein α subunit,Gαs)编码基因GNAS发生体细胞激活突变所致,估计患病率为1/1 000 000~1/100 000[1]。疾病表型复杂,除了可以出现多部位骨骼异常外,还可以有内分泌多器官受累。近年来,研究发现约4%~10%的FD/MAS患者可以合并低磷血症,进一步引发低血磷性佝偻病,不仅导致患者乏力、骨痛,且增加骨折及骨畸形风险,使得骨骼疾病更为复杂、严重,其治疗也极其困难[2]。本文报道1例MAS合并低血磷性佝偻病的罕见病例,分析疾病诊治经过,详细调查患者的临床表现、血生化指标及骨影像学变化,并复习文献,以探讨FD/MAS合并低磷血症的机制及治疗。
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
McCune-Albright综合征(McCune-Albright syndrome, MAS)是一种非常罕见的疾病,其典型临床特点是骨纤维异样增殖症(fibrous dysplasia, FD)、皮肤牛奶咖啡斑和(或)内分泌器官功能紊乱三联征。近期研究表明,MAS的主要发病机制是鸟苷酸结合蛋白的刺激型α亚基(Guanine nucleotide-binding protein α subunit,Gαs)编码基因GNAS发生体细胞激活突变所致,估计患病率为1/1 000 000~1/100 000[1]。疾病表型复杂,除了可以出现多部位骨骼异常外,还可以有内分泌多器官受累。近年来,研究发现约4%~10%的FD/MAS患者可以合并低磷血症,进一步引发低血磷性佝偻病,不仅导致患者乏力、骨痛,且增加骨折及骨畸形风险,使得骨骼疾病更为复杂、严重,其治疗也极其困难[2]。本文报道1例MAS合并低血磷性佝偻病的罕见病例,分析疾病诊治经过,详细调查患者的临床表现、血生化指标及骨影像学变化,并复习文献,以探讨FD/MAS合并低磷血症的机制及治疗。
患者男性,5岁2个月。主诉:发现骨骼畸形2年。患者为第一胎第一产,足月剖腹产,出生体重3 kg,身长不详,人工喂养,1岁会走路,智力及生长速度与同龄儿无异。患者3岁时出现跛行,伴有乏力、右下肢疼痛、右下肢短于左侧及行走缓慢。患者无骨折史,无明显口干、多饮及夜尿增多,否认手足麻木、抽搐,否认肉眼血尿、尿中排石,否认面容改变、手足增大,否认心慌、手抖及大汗,尚未出现变声。二便无异常,精神、睡眠可。既往史:先天性右眼睑下垂,1岁行矫形手术治疗。个人、家族史无殊,父母非近亲婚配,否认类似疾病家族史。体格检查:身高114.5 cm(位于同龄同性别儿0~-1 SD),体重18 kg(位于同龄同性别儿0~-1 SD),蹒跚步态,背部中线偏右侧及右臀部皮肤见大片边界不规则的咖啡斑,右眼睑稍下垂,巩膜不蓝,牙本质未见异常。甲状腺未及肿大,心肺腹无殊。胸骨稍向前突,手镯、脚镯征阳性,未见肋骨串珠,脊柱无侧弯,右下肢短于对侧,为50 cm,左下肢54 cm(异常体征详见图1),胸廓、骨盆及脊柱压痛(-),阴毛Ⅰ期,双侧睾丸体积2 mL。辅助检查:血常规、肝功能、肾功能正常;血磷0.66 mmo/L、血钙2.34 mmol/L。血清碱性磷酸酶(ALP)1 252 U/L、Ⅰ型胶原氨基端前肽(procollagen type Ⅰ N-peptide, PⅠNP)1 148.0 ng/mL,Ⅰ型胶原羧基端肽(β-cross linked C-terminal telopeptide of type Ⅰ collagen, β-CTX)1.78 ng/mL,其中ALP、β-CTX、PⅠNP均较同龄儿明显升高[3,4,5,6],25-羟维生素D(25-OHD)22.1 ng/mL,提示维生素D不足,甲状旁腺素(parathyroid hormone, PTH)34.0 pg/mL;甲状腺功能、胰岛素样生长因子Ⅰ(IGF-Ⅰ)及性激素水平均在正常范围(表1)。骨骼X线片提示右侧股骨及胫骨皮质变薄、骨髓腔扩大,右侧胫骨弯曲畸形,骨纹理结构不清,髓腔呈磨玻璃及囊状扩大表现;四肢长骨骨小梁稀疏,四肢长骨干骺端呈杯口状、毛刷样改变(图2)。采用双能X线骨吸收仪测量的骨密度:腰椎1~4为0.455 g/cm2,Z评分-1.04;股骨颈0.462 g/cm2,Z评分-1.70(表1)。放射性核素骨显像:右下肢短、形态失常,右侧股骨中段及胫骨中下段见放射性摄取增高区,考虑多骨型骨纤维异样增殖症可能(图3)。
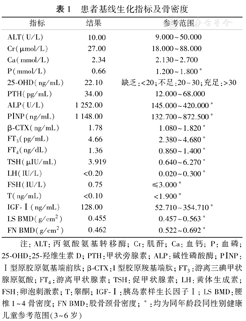
患者基线生化指标及骨密度
患者基线生化指标及骨密度
| 指标 | 结果 | 参考范围 |
|---|---|---|
| ALT(U/L) | 10.00 | 9.000~50.000 |
| Cr(μmol/L) | 27.00 | 18.000~88.000 |
| Ca(mmol/L) | 2.34 | 2.130~2.700 |
| P(mmol/L) | 0.66 | 1.200~1.800* |
| 25-OHD(ng/mL) | 22.10 | 缺乏:<20;不足:20~30;充足:>30 |
| PTH(pg/mL) | 34.00 | 12.000~68.000 |
| ALP(U/L) | 1 252.00 | 145.000~420.000* |
| PⅠNP(ng/mL) | 1 148.00 | 132.700~872.500* |
| β-CTX(ng/mL) | 1.78 | 1.080~1.820* |
| FT3(pg/mL) | 4.66 | 2.380~4.680* |
| FT4(ng/dL) | 1.36 | 0.860~1.400* |
| TSH(μIU/mL) | 3.919 | 0.640~6.270* |
| LH(IU/L) | <0.20 | 0.020~0.300* |
| FSH(IU/L) | 0.75 | ≤3.000* |
| T(ng/mL) | <0.10 | <1.900* |
| IGF-Ⅰ(ng/mL) | 128.00 | 52.710~354.710* |
| LS BMD(g/cm2) | 0.455 | 0.457~0.563* |
| FN BMD(g/cm2) | 0.462 | 0.522~0.692* |
注:ALT:丙氨酸氨基转移酶;Cr:肌酐;Ca:血钙;P:血磷;25-OHD:25-羟维生素D; PTH:甲状旁腺素;ALP:碱性磷酸酶;PⅠNP:Ⅰ型原胶原氨基端前肽;β-CTX:I型胶原羧基端肽;FT3:游离三碘甲状腺原氨酸;FT4:游离甲状腺素;TSH:促甲状腺素;LH:黄体生成素;FSH:卵泡刺激素;T:睾酮;IGF-Ⅰ:胰岛素样生长因子Ⅰ;LS BMD:腰椎1~4骨密度;FN BMD:股骨颈骨密度;*:均为同年龄段同性别健康儿童参考范围(3~6岁)
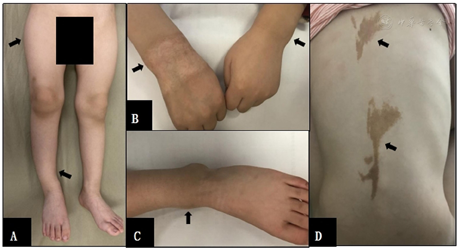
注:A:双下肢大体照片,箭头显示双下肢不等长,右下肢较左下肢短,右下肢弯曲畸形;B:双手手腕大体照片,箭头显示双侧手腕手镯征阳性;C:左脚脚踝大体照片,箭头显示脚镯征阳性;D:背部皮肤大体照片,箭头显示背部中线偏右处皮肤牛奶咖啡斑
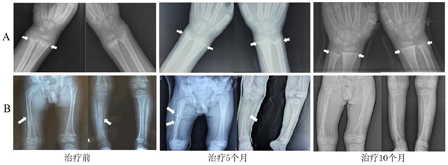
注:A:治疗前后尺桡骨干骺端X线片,箭头显示治疗前尺桡骨骨小梁模糊,干骺端呈杯口状、毛刷样改变;治疗后骨小梁清晰、毛刷状改变减轻;B:治疗前后下肢长骨X线片,箭头显示治疗前右侧股骨及胫骨皮质变薄、骨髓腔扩大,右侧胫骨弯曲畸形、髓腔呈囊状扩大表现;治疗5个月X线示右侧股骨上段骨折;治疗10个月时骨折基本愈合
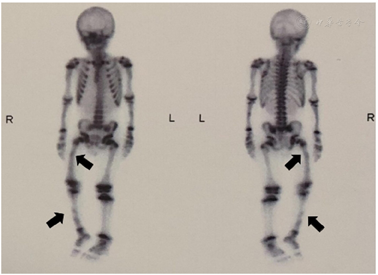
注:箭头示右侧股骨中段及右侧胫骨中下段放射性摄取增高区
诊治经过:针对低血磷性佝偻病,予以骨化三醇0.25 μg/次,每日1次,中性磷合剂(磷酸氢二钠29 g,磷酸二氢钾6.4 g,混匀,加水至1 000 mL)20 mL/次,每天4次服用。治疗2个月后患儿乏力、骨痛减轻,生化检查示血磷上升至0.92 mmol/L,血钙正常,但血PTH水平升高至131.8 pg/mL,β-CTX、ALP及PⅠNP等指标仍有上升,骨化三醇加量至0.25 μg/次,每日3次,中性磷剂量同前,患者骨痛及乏力逐渐改善,活动能力提高。然而,患儿治疗4个月时不慎摔倒,出现右侧股骨骨折,予以石膏固定治疗,继续服用上述药物3个月,血钙磷、PTH水平正常,ALP、PⅠNP明显下降,β-CTX水平仍高。影像学见四肢长骨干骺端骨小梁清晰、毛刷状改变减轻(图2),加用唑来膦酸2 mg静脉输注,以治疗骨纤维异样增殖症,维持原骨化三醇及中性磷剂量。患儿输液后出现发热,持续2 d,体温最高38.3℃,予对乙酰氨基酚混悬液缓解症状,患者无其他明显不良反应。唑来膦酸治疗3个月后随访,血钙磷正常,PTH稍高(95.9 pg/mL),ALP、PⅠNP、β-CTX水平明显下降(表2及图4)。患儿恢复行走能力,无明显骨痛,X线片示右侧股骨上段骨折愈合,右侧股骨及胫骨弯曲畸形大致同前,四肢长骨骨小梁较前清晰,干骺端毛刷状改变减轻(图2)。将中性磷剂量调整为30 mL/次,每天服用4次,骨化三醇0.25 μg/次,每日3次,继续治疗。
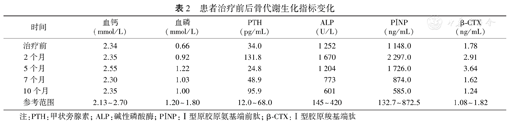
患者治疗前后骨代谢生化指标变化
患者治疗前后骨代谢生化指标变化
| 时间 | 血钙(mmol/L) | 血磷(mmol/L) | PTH(pg/mL) | ALP(U/L) | PⅠNP(ng/mL) | β-CTX(ng/mL) |
|---|---|---|---|---|---|---|
| 治疗前 | 2.34 | 0.66 | 34.0 | 1 252 | 1 148.0 | 1.78 |
| 2个月 | 2.35 | 0.92 | 131.8 | 1 670 | 2 297.0 | 2.91 |
| 5个月 | 2.55 | 1.22 | 24.8 | 1 204 | 1 726.0 | 3.64 |
| 7个月 | 2.30 | 1.03 | 48.9 | 773 | 874.0 | 1.62 |
| 10个月 | 2.35 | 1.00 | 95.9 | 601 | 585.0 | 1.24 |
| 参考范围 | 2.13~2.70 | 1.20~1.80 | 12.0~68.0 | 145~420 | 132.7~872.5 | 1.08~1.82 |
注:PTH:甲状旁腺素;ALP:碱性磷酸酶;PⅠNP:Ⅰ型原胶原氨基端前肽;β-CTX:Ⅰ型胶原羧基端肽
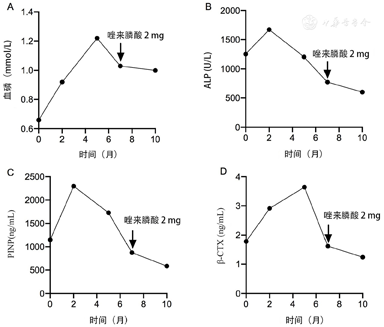
注:A:治疗前后血磷变化情况;B:治疗前后血ALP变化情况;C.治疗前后血PⅠNP变化情况;D:治疗前后血β-CTX变化情况;ALP:碱性磷酸酶;PⅠNP:Ⅰ型原胶原氨基端前肽;β-CTX:I型胶原羧基端肽
本例主要特点如下:幼年起病,慢性病程;临床表现为骨痛、骨骼畸形、步态异常、乏力,皮肤牛奶咖啡斑、右下肢短、右侧股骨及胫骨弯曲畸形;低磷血症,骨转换生化指标ALP、β-CTX及PⅠNP明显升高;X线片示骨纤维异样增殖症及骨软化表现;骨显像示右侧股骨及胫骨放射性增高。经活性维生素D、中性磷治疗后,患者血磷达正常,骨痛及乏力症状减轻,ALP、β-CTX及PⅠNP下降,X线片提示骨软化表现减轻。患儿摔倒后出现右侧股骨骨折,骨折愈合后,给予唑来膦酸治疗FD,患者病情进一步改善。
根据MAS的诊断标准:FD合并≥1个骨外表现,或出现≥2个骨外表现,骨外表现包括特征性皮肤牛奶咖啡斑、非促性腺激素依赖型性早熟、非自身免疫性甲状腺功能亢进症、生长激素过度分泌、新生儿高皮质醇血症及肌内黏液瘤[7]。本例患儿具有多骨型FD及典型皮肤牛奶咖啡斑,考虑MAS诊断明确。
近年来,MAS的发病机制研究有所进展,已经阐明MAS为重要的G蛋白耦联信号通路异常所致,即Gαs的编码基因GNAS发生体细胞激活性突变,导致细胞内环腺苷酸(cAMP)水平升高,G蛋白耦联信号通路持续激活,引发相应临床表现[8,9]。在骨骼,成骨细胞中异常激活的Gαs引起cAMP过度分泌,导致骨髓基质细胞异常增殖,形成纤维性骨组织取代正常骨骼,导致FD;在病变骨骼中,破骨细胞数量及活性明显增加,骨吸收加快[10]。FD可为单骨型或多骨型,常见于颅面骨及四肢长骨,X线表现为骨皮质变薄,病变骨膨胀样改变[11];核素显像可反映病变范围及严重程度,FD病灶呈异常放射性摄取增高。FD患者可有骨转换生化指标的明显升高,包括ALP、PⅠNP、β-CTX及骨钙素等[8]。FD病变活动高峰期常常为6~10岁,可引发骨痛、骨折、骨畸形等并发症,严重影响患儿生活质量[12]。除骨骼外,GNAS突变形成的异常克隆随胚胎生长发育迁移,呈镶嵌型分布于皮肤、内分泌腺体等骨骼外组织中,Gαs异常激活导致多种临床表现,如cAMP分泌增多激活促黑素细胞激素受体通路,导致皮肤局部黑色素分泌增多,出现牛奶咖啡斑;如cAMP依赖的多种内分泌激素分泌增多,包括性腺激素、甲状腺激素及生长激素等,可以引起性早熟、甲状腺功能亢进症、肢端肥大症等[9,13]。
目前对于MAS患者合并的FD,治疗较为困难。研究发现FD病灶中破骨细胞数量及活性增加,抗骨吸收药物可能有一定的效果,双膦酸盐应用相对广泛[14]。研究表明,静脉双膦酸盐治疗可以降低患者骨转换生化指标、减轻骨痛、增加骨密度、减少骨折风险,但难以逆转骨骼膨胀样改变的进展[15,16,17]。近期研究提示核因子κβ受体活化因子配基(receptor activator of nuclear factor κβ ligand, RANKL)的单克隆抗体可能对于FD有效,可使骨转换生化指标显著下降、骨痛减轻,但多为个案报道,其安全性及有效性尚待进一步研究[18]。在应用抗骨吸收药物治疗前,需明确血钙磷、维生素D水平,保证血钙磷正常、维生素D充足。2019年FD/MAS指南推荐在启动双膦酸盐治疗前至少维持血磷正常6个月以上[7],故本例患儿在血磷正常后7个月,方启动双膦酸盐治疗。
本例除FD外,尚有佝偻病体征、低磷血症,影像学提示多部位骨小梁呈磨玻璃样改变,长骨干骺端呈现杯口样及毛刷状改变,提示其存在低血磷性佝偻病。近期研究表明,FD/MAS患者合并低血磷性佝偻病的可能机制为FD病灶过度分泌成纤维细胞生长因子23(fibroblast growth factor 23, FGF23),其下调近端肾小管钠-磷共转运子的表达,抑制肾小管重吸收磷,肾磷排泄增加,且FGF23抑制肾小管1α-羟化酶活性,使1,25二羟基维生素D3[1,25(OH)2D3]合成减少,导致低磷血症[1,19]。低磷血症抑制骨矿化,导致肌无力,进一步加重MAS患者的骨骼病变及活动障碍。一项回顾性研究显示,MAS患儿中血磷低者较血磷正常者骨折频率更高、骨折发生更早[20]。本例患儿为多骨型FD合并低血磷性佝偻病,具有明显骨痛、骨骼畸形、活动障碍及轻微外力下骨折,提示对于合并低磷血症的MAS患者,应更重视对骨骼病变的评估及治疗。根据FD/MAS实践指南[7],对于合并低血磷性佝偻病者,建议采用活性维生素D及中性磷溶液治疗,儿童推荐剂量为活性维生素D 15~60 ng·kg-1·d-1,分2~3次口服,中性磷15~60 mg·kg-1·d-1,分4~5次口服。本例患儿经中性磷及活性维生素D治疗后,骨痛缓解、活动能力提高,影像学提示病变减轻,骨转换生化标志物有显著下降,MAS合并的佝偻病有所改善。血磷水平受饮食、年龄、维生素D营养状况等多因素影响[21],治疗过程中建议监测血钙、血磷、ALP、PTH及24 h尿钙变化,及时调整药物剂量,以防止出现继发甲状旁腺功能亢进和高尿钙症。此外,近年来研究显示,FGF23单克隆抗体Burosumab可特异性抑制FGF23活性,增加肾脏对磷的重吸收及1,25(OH)2D3的合成,在X染色体连锁遗传的低血磷性佝偻病患儿中已取得较好疗效[22],但尚无在MAS合并低血磷性佝偻病中应用的报道,未来值得尝试。此外,MAS患者如有内分泌激素过度分泌,如合并性早熟、甲状腺功能亢进症、肢端肥大症等,也应及早发现,并采取相应治疗措施[12]。
综上,本文报道1例罕见的MAS患者,具有多骨型骨纤维异样增殖症,合并罕见的低血磷性佝偻病,尽早给予中性磷溶液、活性维生素D治疗有助于改善低血磷性佝偻病,在纠正了低磷血症后,给予强有效的骨吸收抑制剂,有助于延缓骨纤维异样增殖症的进展。今后对于罕见病MAS发病机制及治疗的深入研究,有助于提高我们对骨骼及多种内分泌器官的重要调控通路——G蛋白耦联信号通路的认识。
所有作者均声明不存在利益冲突

























