
遗传性血色病(hereditary hemochromatosis,HH)在中国人群中较为罕见,该病可累及多系统,临床表现特异性低,若对其认识不足,极易误诊漏诊。本文报道1例以糖尿病为首诊,而后诊断HH的病例,该患者除肝功能异常外,还出现性腺功能减退和骨质疏松等多种内分泌代谢性疾病;同时结合文献探讨HH的临床特征、诊治方法及其与内分泌功能障碍的联系,以提高临床医师对该病的认识。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
血色病是由铁代谢紊乱导致的体内铁超载的一类疾病,因过多的铁沉积在肝脏、胰腺、心脏、关节和其他器官而导致广泛的组织纤维化、器官结构和功能损伤[1]。该病在我国临床上较为罕见。本文报道1例经影像学检查和肝脏、骨髓组织穿刺活检考虑为遗传性血色病(hereditary haemochromatosis, HH)的病例,患者除肝功能异常外还出现了继发性糖尿病、性腺功能减退和骨质疏松等多内分泌代谢异常。同时结合文献探讨HH的临床特征、诊治方法及其与内分泌功能障碍的联系,为临床医师对该病的诊断提供参考。
患者男,66岁,于2020年8月和2021年5月先后2次入住我科。患者2020年5月出现口干、多饮、多尿,3个月内体重下降约5 kg,至当地医院就诊,测随机血糖37.5 mmol/L,予以胰岛素降糖治疗后症状好转出院,院外未监测血糖、未坚持降糖治疗;2020年8月上述症状加重,伴精神困倦、纳差1周就诊于本院门诊后收治入科。HbA1C 15.4%(正常参考范围3.9~6.1,下同)、空腹C肽0.094 nmol/L(0.3~1.3)、餐后2 h C肽0.231 nmol/L,胰岛自身抗体阴性,初步考虑为成人隐匿性自身免疫性糖尿病,予以基础联合餐时胰岛素降糖,但查体发现患者皮肤呈青铜色,检验提示血清铁45.6 μmol/L(10.6~28.3)、铁蛋白>2 000.00 ng/mL(21.81~274.66),头颅磁共振成像(magnetic resonance image, MRI)提示有异常信号,腹部MRI提示肝脏及胰腺信号减低,完善肝组织病理检查后诊断"血色病继发性糖尿病"。
既往史:既往体健,否认血液病、肝炎病史、否认长期服用铁剂史,无手术史、外伤史,无输血史,既往吸烟45年余,1包/日,现已戒烟1年,偶饮酒,无酗酒史。
家族史:否认血色病及糖尿病家族史,但患者女儿可见明显皮肤色素沉着,由于经济问题未行相关检查。
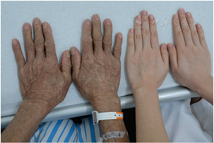
查体:体温36.4℃,脉搏70次/min,呼吸20次/min,血压118/66 mmHg(1 mmHg=0.133 kPa),身高159 cm,体重50 kg,体重指数19.77 kg/m2。心肺(-),腹平软,无压痛、反跳痛,肝脾未触及,肠鸣音正常。四肢皮肤干燥,无肝掌及蜘蛛痣,全身皮肤有青铜色色素沉着(图1),四肢关节无红肿、压痛、畸形。实验室检查:门冬氨酸氨基转移酶90 U/L(0~40)、丙氨酸转氨酶62 U/L(0~50)、血红蛋白136 g/L(120~165)、红细胞4.17×1012/L(4.3~5.8×1012)、白蛋白38.3 g/L(40~55)、脂肪酶41 U/L(18~180)、淀粉酶69 U/L(24~151)、8:00皮质醇767.50 nmol/L(171~536)、8:00促肾上腺皮质激素(adrenocorticotropic hormone, ACTH) 11.340 pmol/L(1.6~13.9)、16:00皮质醇353.40 nmol/L、16:00 ACTH 2.450 pmol/L、0:00皮质醇240.70 nmol/L、0:00 ACTH 0.883 pmol/L、睾酮1.97 nmol/L(6.07~27.1)、垂体催乳素7.66 μg/L(2.64~13.13)、FSH 2.40 IU/L(1.27~19.26)、LH 2.43 IU/L(1.24~8.62)、尿微量白蛋白11.8 mg/L(<30)、尿微量白蛋白/肌酐20.4 mg/g(<30),肝炎病毒抗体、男性肿瘤标志物均为阴性。
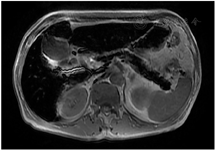
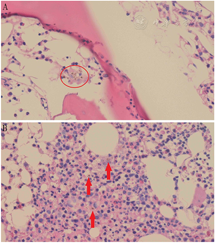
影像学检查:头颅MRI(图2)提示双侧额颞顶枕叶皮质下及基底节区低信号,考虑代谢性脑病可能。腹部MRI(图3)提示肝实质和胰腺T1WI及T2WI+FS信号弥漫性减低,均呈极低信号。腹部超声提示肝脏轻度纤维化、肝左叶体积增大,脾轻度肿大,胰腺萎缩伴纤维化、双肾体积缩小、前列腺体积缩小。睾丸超声提示双侧睾丸萎缩。骨密度提示左侧股骨颈骨密度(bone mineral density, BMD)测量值符合骨质疏松(腰椎L1-4 BMD为0.879 g/cm2,T值-1.7,Z值-0.5;左股骨颈BMD为0.638 g/cm2,T值-2.6,Z值-1.2;左股骨上端为0.731 g/cm2,T值为-2.0,Z值为-1.4)。心电图示窦性心动过缓、异常"Q"波。
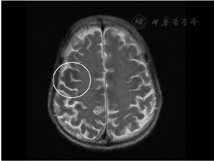
注:白色圈内:颞叶低信号区
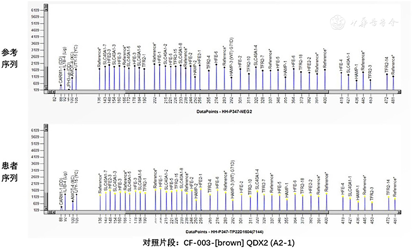
肝组织病理(图4、图5):胆管细胞及汇管区均可见含铁血黄素,铁染色,结合病史考虑遗传性血色病可能性大。骨髓组织病理(图6):少许含铁血黄素沉积(细胞外铁:阳性;细胞内铁:89%阳性)。基因检测(图7):未检测到1/2/3/4型血色病相关基因外显子编码区的致病变异。
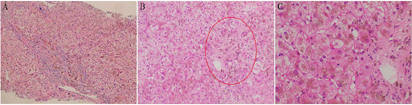
注:A:×40倍;B:×100倍;C:×200倍
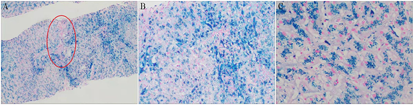
注:A:×40倍;B:×100倍;C:×200倍
最终诊断:(1)原发性血色病(①继发性糖尿病,②肝功能异常,③性腺功能减退症,④骨质疏松症,⑤代谢性脑病);(2)窦性心动过缓;(3)异常Q波。
诊疗经过:患者未接受静脉放血疗法,未遵嘱购买服用雄激素,院外坚持基础联合餐时胰岛素降糖,复方甘草酸苷护肝,碳酸钙维D3、骨化三醇补钙等对症治疗,并减少膳食铁的摄入和避免接触铁器。2021年5月入院复查时血清铁为27.9 μmol/L,较前下降,但铁蛋白仍高(铁蛋白>2 000.00 ng/mL、稀释后铁蛋白5 391.39 ng/mL),空腹血糖控制在8 mmol/L以内,餐后2 h血糖10 mmol/L以内,肝功能较前恢复(门冬氨酸氨基转移酶63 U/L、丙氨酸转氨酶56 U/L),但骨质疏松有所加重(左股骨颈BMD为0.590 g/cm2,T值为-3.0,Z值为-1.6;左股骨上端为0.685 g/cm2,T值为-2.4,Z值为-1.8)。
临床上血色病通常因实验室检查提示血清铁蛋白或转铁蛋白饱和度升高而发现,但上述两项指标特异性较低,往往需要进一步检查来明确诊断[2]。影像学是诊断血色病的重要辅助检查,当肝脏、脾脏和胰腺等脏器出现铁超载时,因铁的超顺磁性效应,MRI-T2加权序列可表现出明显低信号[3]。若肝实质内铁含量>7 mg/g肝重量时,MRI-T2序列图像可出现"黑肝症",这对量化和可视化血色病患者肝脏铁含量具有重要意义[4]。本例患者MRI-T2序列可见肝实质信号弥漫性减低,表现为明显"黑肝症",除此之外胰腺和双侧额颞顶枕叶皮质下及基底节区也出现信号缺失,提示本例患者肝脏、胰腺,甚至是脑部均可能存在铁超载。
血色病根据病因可分为继发性血色病(secondary hemochr-omatosis,SH)和HH两类,前者是其他疾病或治疗措施导致体内铁超载(如先天性或获得性慢性贫血、长期口服铁剂、反复输血、慢性肝病和恶性肿瘤等)[5];后者是由于第6号染色体上的基因突变导致小肠上皮细胞对食物中铁吸收增加而造成的先天性铁代谢障碍,是白种人中最常见的常染色体隐性遗传疾病,尤其在北欧高加索和日耳曼人群中,其发病率达1/250~1/220,而在亚裔人群中相对少见[6]。本例患者既往体健,无贫血、长期口服铁剂和输血史,肝炎病毒相关抗体阴性,因此考虑原发可能性大。
1996年Feder等[7]发现稳态铁调节基因(homeostatic iron regulator gene, HFE)突变是大多数HH患者的病因所在,目前基因检测已成为诊断HH的金标准。迄今为止,大多数HH按照基因突变情况可分为4个类型[8]:1型为HFE基因相关型,最常见的是C282Y纯合子基因突变;2、3、4型为较少见的非HFE相关型,包括铁调素调节蛋白基因突变导致的青少年血色病(2A型)、肝脏抗菌蛋白基因突变(2B型)、转铁蛋白受体-2基因突变(3型)、铁转运蛋白基因(SLC40A1)突变(4型)。国人基因突变模式与欧美人热点突变显著不同,尽管本例患者经基因检测未发现HH1/2/3/4型相关基因外显子编码区的致病变异,但亦不能排除存在其他罕见基因位点变异的可能性。而明确诊断血色病的类型可指导临床治疗,HH的一线治疗方案为静脉放血术[9]。我们建议患者行放血治疗,患者及家属未能接受,遂嘱其避免进食红肉和减少接触铁器,8个月后复查血清铁含量有所下降,但铁蛋白水平仍高。
目前肝活检的主要用途是对纤维化进行分期,常用的组织染色法包括苏木精-伊红(hematoxylin-eosin, HE)和普鲁士蓝染色[5]。肝脏活检也曾用于鉴别HH和SH,HH典型的肝组织病理学特征为铁沉积优先发生在门静脉区(腺泡1区)的细胞内,并向周围减少,表现出明显的沉积梯度,并随着疾病进展,胆管上皮细胞内铁沉积和汇管区纤维化程度将加重[10]。本例患者肝组织病理虽无明显铁沉积梯度变化,但HE及普鲁士蓝染色均可见胆管细胞及汇管区有大量含铁血黄素沉积,汇管区扩大及纤维组织增生(图4B、图5A),提示患者病程长,病变重。
HH早期通常无症状,随着铁沉积的进展广泛性累及到全身多个系统,可出现多种临床症状,诊断具有一定难度[11]。青铜色皮肤色素沉着、肝硬化和糖尿病是HH典型的临床三联征[12]。肝脏是HH最常累及的器官,其临床表现具有多样性,例如肝脏转氨酶升高、右上腹疼痛、肝肿大、肝纤维化,晚期甚至可进展为肝癌,而吸烟、饮酒等不良生活习惯则会加重此类患者的铁超载[5]。本例患者转氨酶升高,腹部超声和肝组织活检提示已出现肝纤维化,考虑为患者多年的吸烟史、饮酒史加剧铁超载所引起的病理变化。
HH患者的胰腺内铁超载常导致合并糖耐量受损或糖尿病,早期研究认为HH患者的糖尿病患病率约在30%~60%[13],但在发现基因突变与HH的关系并因此正确诊断和及时治疗后,患病率下降至13%~23%[14]。尽管HH诱发糖尿病的机制尚未完全阐明,但目前认为与胰腺铁超载导致的毒性对胰岛β细胞的损伤[15],以及与肝脏铁超载致胰岛素受体损伤而引起胰岛素抵抗有关[16]。且研究表明,对于HH继发性糖尿病患者,若血糖控制不佳,微血管并发症的发生率会更高[17]。本例患者胰岛功能差,结合MRI提示胰腺有弥漫性信号减低,考虑糖尿病与大量铁沉积导致胰岛β细胞功能受损关系密切,患者目前使用基础联合餐时胰岛素降糖治疗,血糖控制良好。幸运的是,患者眼底检查未见糖尿病视网膜病变,实验室检查提示糖尿病肾病无明显进展。此患者淀粉酶、脂肪酶水平正常,无脂肪泻,目前疾病进展暂未对胰腺外分泌功能产生影响。
性腺功能减退症也是HH患者常见的内分泌腺疾病,其病理机制为垂体前叶促性腺激素细胞内铁沉积,从而引起促性腺激素分泌不足,导致继发性性腺功能减退症,在男性,可表现为体毛减少、性欲减退、阳痿、不同程度的睾丸萎缩[18],女性通常会出现月经排卵异常、不孕、绝经期提前和性欲减退等症状[19]。本患者睾酮低于正常水平,促性腺激素也接近低值,影像学检查提示睾丸萎缩,考虑已出现继发性性腺功能减退症。HH患者的性腺功能低下可促进骨质疏松的发生发展。研究表明HH患者骨质疏松患病率为25%~34%,骨量减少的患病率约40%~79%[19]。其发病机制一方面是铁超载导致的氧化应激、慢性炎症状态抑制成骨细胞的增殖和活性[20],另一方面是性腺功能低下不能抑制破骨细胞活性,导致骨吸收增加,且既往研究发现,HH患者的股骨BMD比颈椎、腰椎下降更明显[21]。因此本例患者的骨质疏松症考虑与其铁超载和性腺功能减退有关。若铁超载累及甲状旁腺导致甲状旁腺功能障碍,也会加重骨质疏松[19],但本患者未见甲状旁腺功能减退。HH致骨质疏松的治疗关键是处理原发病,即通过静脉放血术减少铁沉积,并改善激素分泌障碍,辅以钙剂和维生素D对症治疗,越早治疗预后越好[19]。但本例患者仅以补钙对症处理,原发病尚未明显好转、未接受雄激素替代治疗性腺功能减退症,可能是其左股骨颈和股骨上端BMD下降的重要原因。
此外,HH患者可合并甲状腺和肾上腺功能障碍,机制可能是铁沉积直接出现在甲状腺、肾上腺本身,或是沉积于垂体前叶的促甲状腺素细胞或促肾上腺素细胞内,导致继发性甲状腺、肾上腺功能减退症,但垂体内此类细胞中的铁沉积较促性腺激素细胞少,因此HH合并上述疾病在临床上较少见[17]。本例患者的实验室检查也提示无明显甲状腺和肾上腺功能减退症。
总之,HH是我国临床上罕见的铁代谢缺陷性疾病,早期可无任何症状,发现时往往已累及肝脏、关节、心肌、内分泌腺等多个脏器,临床表现多样但特异性低,容易漏诊、误诊;而本例患者除肝功能异常外,则以糖尿病、性腺功能减退和骨质疏松症等内分泌代谢疾病为主。因此,当临床上遇到内分泌功能障碍合并铁蛋白水平升高的患者时,应仔细考虑HH的可能性;对于已确诊HH的患者,则需全面评估内分泌功能以明确受累腺体并尽早制定个体化的治疗方案。
所有作者均声明不存在利益冲突

























