
线粒体脑肌病伴高乳酸血症、卒中样发作综合征(mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes, MELAS)是最常见的原发性线粒体疾病之一。本文报道了1例罕见的由m.3252A>G(MT-TL1基因)突变导致的MELAS综合征。先证者为6岁女童,以反复抽搐和高乳酸血症为主要表现,线粒体DNA(mtDNA)基因测序示患儿及其母亲和外婆均携带m.3252A>G(MT-TL1基因)变异,该变异在患儿外周血和尿沉渣中的异质率分别为66.53%和97.42%,明显高于母系亲属。结合国外已报道的3个家系资料,该变异评级为致病性变异。通过复习国内外文献,探讨了MT-TL1基因致病变异的基因型-表型相关性。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
线粒体疾病是一组由核DNA(nuclear DNA, nDNA)或线粒体DNA(mitochondrial DNA, mtDNA)变异导致氧化磷酸化功能障碍为特征的一组遗传性疾病[1]。流行病学研究表明,线粒体病整体最低患病率为1/5 000[1]。线粒体脑肌病伴高乳酸血症、卒中样发作综合征(mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes, MELAS)是线粒体病中最常见的综合征之一,90%病例由线粒体亮氨酸转运核糖核酸1(MT-TL1)基因突变引起,其余10%由其他mtDNA或nDNA致病变异引起[2,3]。本研究对本院2020年1月收治的1例由m.3252A>G(MT-TL1基因)变异导致MELAS综合征的患者进行报道,并通过文献复习,对已报道的MT-TL1基因致病变异进行梳理,以期加深对该基因变异的基因型-表型相关性的认识,从而指导临床。
患儿女性,6岁1个月,因"半天内抽搐4次,伴呕吐1次"入院。表现为全身强直阵挛性发作,持续约1 min后自行缓解,间隔数分钟再次发作,共4次,伴高乳酸血症。患儿为第2胎第2产,出生史正常,生后智力、运动发育均落后(17个月会叫妈妈,26个月会走路,4岁会说简单句子)。患儿母亲有糖尿病,父亲体健,非近亲婚配,有1个姐姐,12岁因"酸中毒"死亡。体格检查:身高108 cm(-2 SD),体重15.5 kg(<-2 SD),神志清,双眼外眦上斜,前额低平,前额发际线低,毛发茂盛,分布正常。四肢肌力Ⅴ-,肌张力正常,腱反射可引出,双侧巴宾斯基征阳性,余无明显异常。辅助检查:血乳酸4.8~9.8 mmol/L(参考范围0.5~2.5 mmol/L,下同);心肌型肌酸激酶同工酶167.1 ng/mL(0~3.61 ng/mL);血生化肌酸肌酶282.4~757.6 U/L(25~225 U/L)、天冬氨酸转氨酶162.9 U/L(10~67 U/L)、丙氨酸转氨酶40.7~48 U/L(5~35 U/L)。血糖、血氨、遗传代谢疾病筛查正常。头颅磁共振(MRI)成像:双侧放射冠、豆状核异常信号。定期复查头颅影像学检查发现病灶缓慢进展(图1)。心电图:心室预激。心脏超声:左室肌非对称性增厚伴回声稍增强,左室心内膜局部增厚。
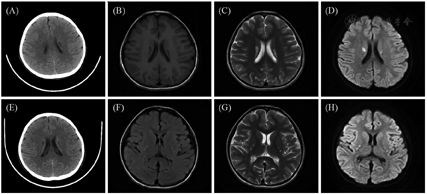
注:(A)~(D)先证者首诊时的影像学检查;(E)~(H)先证者6岁11个月复诊时的影像学检查;(A)计算机断层扫描(CT)显示两侧侧脑室旁卵圆形低密度灶;(B)~(D)磁共振成像(MRI)显示双侧放射冠对称性条带状异常信号,长T1长T2信号,DWI高信号影;(E)CT显示两侧侧脑室旁小片状低密度灶,较前略进展;(F)~(H)MRI显示双侧豆状核、尾状核体部见小斑片状,长T1长T2信号,DWI呈高信号
与患儿家属沟通并经过苏州大学附属儿童医院伦理委员会批准(批准文号:2021CS003)后,行基因检测。收集患儿及其母亲、外婆及舅舅外周血、尿液提取DNA,均用长程PCR/大规模并行测序(LR-PCR/MPS)方法进行mtDNA测序。流程如下:利用受检者基因组DNA,使用特异性引物进行mtDNA全长扩增,扩增产物片段化后构建基因组DNA文库(NEB #E7370L),通过下一代高通量测序仪(Illumina)进行测序,测序数据运用NextGene V2.3.4软件比对到美国国家生物技术信息中心(NCBI)数据库提供的mtDNA NC_012920.1参考序列上,对筛查出的变异进行过滤,并添加Mitomap数据库注释信息。在患儿MTTL1基因中检测到m.3252A>G变异,其异质率:外周血为66.53%、尿液为97.42%;其母m.3252A>G变异的异质率:外周血为13.25%、尿液为25.08%;其外婆:外周血为1.60%、尿液为9.43%;舅舅的血和尿中未检测到该变异(图2)。治疗经过:给予鸡尾酒疗法;精氨酸改善血管内皮细胞功能;左乙拉西坦治疗癫痫;卡托普利、螺内酯减轻心脏负荷,预防心室重构;地高辛加强心肌收缩功能;并给予营养支持治疗。住院期间患儿未再抽搐,病情稳定后长期门诊随诊。1年来,仍有反复抽搐,伴头痛、呕吐等症状。
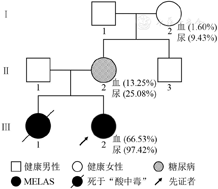
注:MT-TL1:线粒体亮氨酸转运核糖核酸1; MELAS:线粒体脑肌病伴高乳酸血症、卒中样发作综合征
MT-TL1基因为MELAS的常见致病基因,位于mtDNA重链,由76个核苷酸组成,负责编码转运RNA(tRNA) Leu(UUR),后者通过识别密码子UUG和UUA携亮氨酸至延伸的多肽链,参与线粒体蛋白的合成[2,3]。由于MT-TL1基因的核苷酸序列高度保守,单一点突变就可能引起tRNA前体成熟障碍、碱基修饰缺失,进而破坏tRNA Leu(UUR)的构象稳定,降低其氨基酰化水平,导致线粒体内不稳定多肽链增多,以复合体Ⅰ或Ⅳ为主的线粒体蛋白功能受损而致病[3,4]。MT-TL1基因变异致病机制不仅仅局限于氧化磷酸化障碍导致ATP供能不足,引起大脑、肌肉等高代谢器官率先出现能量耗竭,还可引起线粒体损伤后激活的应激反应[5,6]。不同程度线粒体损伤可能激活不同的逆行信号通路,后者可以是线粒体介导的钙稳态、活性氧簇(ROS)和膜电位的变化[7]。MT-TL1基因变异的表型谱分布广,从无症状携带到致死性脑肌病均可发生[1,2,7]。其中,MELAS综合征属于较严重的表型。MELAS综合征目前暂无有效治疗方案,多以支持疗法改善症状,如补充呼吸链组分及上游物质(辅酶Q10、硫辛酸、左卡尼丁、维生素B1);去除蓄积的有害代谢产物(降乳酸、抗氧化);促进血管扩张(精氨酸补充一氧化氮);对症治疗(抗癫痫、控血糖、增强肌肉锻炼)等。正因MELAS发病机制复杂,治疗手段匮乏,故从出现神经缺陷开始,患者总体中位存活时间仅为16.9年,幼年型MELAS相比成年型MELAS有更高的死亡风险[8]。
迄今已报道的m.3252A>G(MT-TL1基因)有3例[4,9,10]。首例由Morten等[9]报道,先证者为13岁女性,以线粒体脑肌病合并色素视网膜病变、痴呆、甲状旁腺功能减退和糖尿病为表型,突变异质率:肌肉为76%,外周血为30%。先证者母亲以卒中样发作和痉挛性瘫痪为表型,肌肉异质率为50%。第2例由O′Callaghan等[10]报道,以MELAS综合征,伴视力下降、听力障碍和认知延迟为表型的13岁女性患者,外周血异质率为42%。第3例由Wong等[4]报道,以偏头痛、癫痫、卒中样发作为表型的16岁男性患者,外周血异质率为42%。本研究中,先证者为6岁女童,临床以MELAS综合征为主,伴智力、运动发育落后,合并心室肌肥厚。应用Wong等[4]的线粒体tRNA变异致病性分类标准,可将m.3252A>G(MT-TL1基因)评为致病性变异(pathogenic),证据包括PS5、PM8、PM9、PM10、PP4和PP7(证据等级:PS表示强致病性证据,PM表示中等致病性证据,PP表示辅助证据)。Wong等[4]的线粒体tRNA变异分类标准与美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics, ACMG)指南[11]对m.3252A>G的致病性评价有区别。根据ACMG指南[11],m.3252A>G的评级为可能致病性变异,证据包括PS4_moderate、PM2、PP3、PP4(证据等级:PS4表示中等强度)。该突变破坏线粒体Ⅰ的活性,引起糖代谢紊乱和ATP产能不足,可导致MELAS综合征[9]。m.3252A>G患者从发病到死亡时间为5~27年(平均15.3年)[4,9,10],预后不佳。本例患者发病年龄小,除具有与既往病例相似的卒中样发作、智力低下症状外,还患有预激综合征和心肌病。该患儿症状重可能与其携带m.3252A>G变异的异质率高相关。
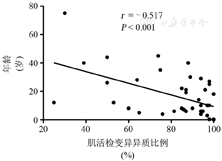
为进一步探讨MT-TL1基因型与表型的关系,本研究以"MT-TL1 mutation""MT-TL1 variant""tRNA Leu(UUR)"为关键词检索NCBI、PubMed、OMIM、中国知网和万方数据等数据库,对MT-TL1基因中已报道的变异位点进行致病性分析。因热点突变"m.3243A>G(MT-TL1基因)"报道数量多,本文只引用相关综述,不展开对m.3243A>G的讨论[2]。本研究共收集到64篇文献,30个变异位点。根据Wong等[4]的mt-tRNA基因变异致病性分类标准重分析,其中符合致病性变异的有15个:A3243G、A3243T、A3251G、A3252G、G3255A、C3256T、T3258C、A3260G、T3271C、A3274G、A3280G、C3287A、T3291C、A3302G、C3303T;符合可能致病性变异的有7个:G3242A、G3244A、G3249A、3271delT、T3273C、G3283A、A3288G。若根据ACMG指南[11]分类,上述30个变异位点中致病性变异10个,可能致病性变异9个[4]。以变异致病性在可能致病性变异等级及以上,并且具备详细临床资料为条件,对收集的64篇文献进行筛选,最终有34篇,共计59例患者符合进一步分析的条件[4,9,10,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33]。分别整理报道中患者性别、发病年龄、首发症状、主要临床表现和突变异质水平的数据资料。59例患者中,男性21例,女性36例,未描述性别1例。描述起病年龄的有42例,通过对发病年龄与肌活检中变异异质水平的Pearson相关性分析,发现两者呈中度负相关,差异有统计学意义(图3)。在对MT-TL1基因的表型进行梳理后,按病情的严重程度进行分级,以患者是否因该变异死亡、是否累及多个系统以及病程进展速度,分为危重型、重型、普通型和轻型(图4)。危重型为致死性的、快进展的且合并其他器官功能衰竭的患者[4,9,10,12,13,14,15,16,17,18,19,20];重型为临床症状较重的脑肌病、脑病、肌病,合并其他系统功能异常[9,17,21,22,23,24,25,26,27];普通型为症状轻的脑肌病和全身性肌病[26,28,29,30,31];轻型为局灶性的肌病、糖尿病、甲状腺功能减退症等[4,16,21,24,26,32,33]。有17个变异在肌肉组织中为异质性,异质率为25%~98%。有4个变异为同质性(G3242A、A3288G、A3302G和C3303T),均以肌病或心肌病为主要表现。以中枢神经系统症状为主的致病变异有12个:A3243T、G3244A、G3249A、A3252G、G3255A、C3256T、T3258C、T3271C、3271delT、A3274G、C3287A和T3291C。总体上异质率高的变异多表现为严重的脑病、脑肌病,随异质率下降可表现为肌病或眼肌病。其中,G3244A、A3252G、C3256T、T3258C、T3271C、3271delT和T3291C变异均有MELAS综合征的表型。G3244A、T3258C、T3271C和T3291C突变会引起tRNA leu(UUR)构象改变,第34位尿嘧啶的5-牛磺酸甲基缺失而影响tRNA leu(UUR)解码能力,导致线粒体编码的呼吸链亚基中不稳定多肽链增多[34]。剩余的A3252G和C3256T变异是否有相同机制,有待进一步研究。以心肌病或肌病为主要症状的变异有9个:G3242A、A3251G、A3260G、T3273C、A3280G、G3283A、A3288G、A3302G和C3303T,其异质率和疾病严重程度的相关性不明显。如A3260G家系中异质率为87%~89%,可表现为单纯肌病或严重的心肌病[25];C3303T异质率为98%时,心肌病患者临床表现轻重不一[20,31,33]。猜测可能与组织nDNA背景及环境等因素的影响有关。
注:MT-TL1:线粒体亮氨酸转运核糖核酸1;未包括m.3243 A>G

注:MT-TL1:线粒体亮氨酸转运核糖核酸1;横轴表示21个突变位点,纵轴表示异质率;黑色为危重型,红色为重症,蓝色为普通型,绿色为轻型,橙色为同一异质率有轻症到重症多个表型;圆形表示肌肉活检,正方形表示尿沉渣;DD:生长发育迟缓;HCM:肥厚型心肌病;RTA:肾小管性酸中毒;ENC:脑病;SHL:感觉神经性听力丧失;GHD:生长激素缺乏症;PEO:进行性眼外肌麻痹;EI:运动耐量不足;MELAS:线粒体脑肌病伴高乳酸血症、卒中样发作综合征;KS-like:PEO、小脑共济失调、肌病色素视网膜病变;MYO:线粒体肌病;DEM:痴呆;RF:肾功能衰竭;MERRF:肌阵挛性癫痫伴随红纤维病;KSS:Kearns Sayre综合征;PSY:精神障碍;DM:糖尿病;CMYO:心肌病;HF:心力衰竭;RSF:呼吸衰竭;CEA:小脑萎缩;ATX:共济失调;WPW:WPW预激综合征
综上,本研究患者为国内首例报道的m.3252A>G(MT-TL1基因)变异导致的MELAS综合征。本案例存在的心脏功能和结构异常在既往文献中未曾报道,丰富了该变异的表型谱。MT-TL1基因变异的异质率水平与患者发病年龄呈负相关,且在累及中枢神经系统的变异中,异质率越高则临床表型越重。
姑苏卫生人才计划(GSWS2020046, GSWS2019051);苏州市科技计划民生科技(SS202064, SS202091);苏州市临床医学中心(Szlcyxzxj202105)
所有作者均声明不存在利益冲突

























