
IgG4相关疾病(immunoglobulin-G4 related disease, IgG4-RD)是由免疫介导的慢性炎症伴纤维化疾病。现报道1例误诊为颅内感染多年的IgG4-RD病例,患者经多次手术治疗症状无改善,病变累及多个系统,实验室检查示多项免疫相关指标异常,IgG4明显升高,且脑组织病理检查示较多淋巴细胞和大量IgG4+浆细胞浸润,确诊为IgG4-RD,予甲泼尼龙冲击治疗,患者症状好转。IgG4-RD是一种几乎可累及人体各个器官的多系统性疾病,临床表现呈多样性,极易误诊、漏诊,临床医师应高度关注该疾病。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患者男,63岁,因"间断发热1年"于2020年6月29日入河南大学人民医院。患者1年前无明显诱因下出现发热,体温最高达38.0 ℃,伴有反应迟钝、吞咽困难、饮水呛咳、颈部硬肿,无咳嗽、咳痰,无胸闷、呼吸困难。患者于外院查血常规示感染相关指标高,CT检查示颅内感染可能(具体不详),予"美罗培南、万古霉素"抗感染治疗,体温可降至正常,但很快反复;后多次因"颅内感染"住院,予抗感染治疗,疗效差。4月余前因颅内高压、脑积水于外院行脑室-腹腔分流术。3月21日外院头颅MRI检查示左侧枕部颅板下积液、左侧颞枕顶叶明显水肿,增强后明显强化;颅脑术后改变。5月21日肺部CT检查示双肺多发结节影、条索影,考虑慢性炎症。为求进一步诊治来河南大学人民医院,门诊以"发热待查:颅内感染?脑室-腹腔分流术后"收入感染科。患者自发病来,神志欠清,精神差,食欲及睡眠可,二便正常,体质量较前无明显减轻。既往史:2003年因颈椎破坏行植骨术;2013年因"发热伴头痛"发现颅内病变,曾就诊于多家医院均未明确诊断,病情缓慢进展;2018年因"头痛加重"行颅内病变切除术,术后考虑感染性病变,间断抗感染治疗,病情仍无改善。体格检查:体温为36.8 ℃,脉搏为88次/min,呼吸为20次/min,血压为135/85 mmHg(1 mmHg=0.133 kPa)。神志欠清,反应迟钝,对答欠切题,计算力下降。双侧额纹变浅,闭目力差,双侧鼓腮力、咀嚼力差;伸舌困难,舌不能完全伸出;双侧瞳孔等大等圆,眼球活动度差,尤其上视、右视时明显,对光反射迟钝,视力下降;颈部红肿、僵硬,无压痛;双肺听诊呼吸音清,未闻及干、湿啰音;心前区无隆起,心界不大,律齐,各瓣膜听诊区未闻及心脏杂音及心包摩擦音;腹部平软,无压痛,肝脾肋下未及,移动性浊音阴性,肠鸣音正常;双下肢无水肿,肌力3级,腱反射(-),病理征(+)。考虑为免疫性疾病,予补液、营养支持等对症治疗。实验室检查:IgG为37 910 mg/L,抗髓过氧化物酶抗体为24.21 RU/mL,IgE为208.3 IU/mL,IgG4为10 400 mg/L;IL-6为115.88 ng/L;ESR为106 mm/1 h;CRP为146.36 mg/L;降钙素原为0.12 μg/L;血常规、尿常规、粪便常规、游离三碘甲腺原氨酸、游离甲状腺素、促甲状腺激素、T.SPOT.TB检测、β-D-葡聚糖试验、半乳甘露聚糖抗原试验、EB病毒DNA、CMV DNA、布鲁菌抗体、肿瘤标志物均阴性。6月30日胸部CT检查示双肺多发结节,见图1。7月2日头颅MRI平扫+增强检查示脑内多发异常信号影;后纵裂池处脑膜、小脑幕及左侧颞部硬脑膜增厚并明显强化;左侧枕叶局部脑穿通畸形形成,见图2。进一步行腰椎穿刺脑脊液检查示单核细胞计数为66×106/L,蛋白质为6.02 g/L,葡萄糖为2.02 mmol/L,氯化物为119.9 mmol/L。考虑为IgG4相关疾病(immunoglobulin-G4 related disease,IgG4-RD)。7月2日请病理科会诊外院术中脑组织病理切片,结果示(左枕)纤维组织增生伴玻璃样变性,局部胶原化,淋巴袖套结构可见,血管周围及硬化间质内见较多淋巴细胞及浆细胞浸润,浆细胞形态成熟,见图3;免疫组织化学染色示纤维及血管周边可见多量IgG4+浆细胞浸润,IgG4+浆细胞约为50~100个/高倍视野,见图4。根据患者检查结果和多科室专家会诊,诊断为IgG4-RD,累及中枢、肺部、皮肤、骨骼。由于患者病变侵犯脑干,有脑水肿、颅内压升高等表现,可能出现呼吸、心跳骤停等严重并发症,与患者家属沟通患者病情。7月10日开始予甲泼尼龙(500 mg,1次/d,静脉滴注)冲击联合丙种球蛋白(20 g,1次/d,静脉滴注)治疗。当天患者眼球转动基本正常,进食无呛咳,意识、反应和言语明显好转,颈部硬肿好转。予静脉滴注甲泼尼龙500 mg 3 d、240 mg 3 d、120 mg 3 d、80 mg 3 d、40 mg 3 d,以及静脉滴注丙种球蛋白20 g (1次/d) 5 d,患者症状明显改善,复查IgG、IgG4、ESR、CRP均较前下降。7月13日肺部CT检查示双肺结节明显缩小,见图5。7月19日头颅MRI检查示颅内局部脑水肿减轻,后纵裂池处脑膜、小脑幕及左侧颞部硬脑膜病变缩小,见图6。患者症状逐渐好转,7月24日患者及家属要求出院。出院后继续予甲泼尼龙治疗,后联合吗替麦考酚酯,患者一般情况稳定,定向力、记忆力、认知力正常,言语流利。
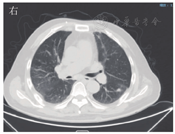
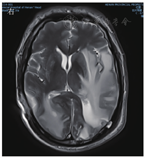


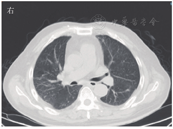
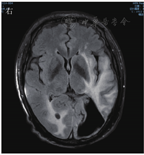
IgG4-RD是一种由免疫介导的慢性炎症伴纤维化疾病,主要组织病理表现为以IgG4+浆细胞为主的淋巴细胞、浆细胞浸润,并伴有席纹状纤维化、闭塞性静脉炎及嗜酸性粒细胞浸润[1,2]。2003年,Kamisawa等[3]首次提出IgG4-RD的概念。2010年,Takahashi等[4]对其正式命名。IgG4-RD好发于中老年男性,几乎可以累及人体的各个器官[5,6]。受累器官因纤维化、慢性炎症等出现增生肿大,从而导致相应压迫阻塞症状或功能障碍[7]。
IgG4相关肥厚性硬脑膜炎(immunoglobulin-G4-related hypertrophic pachymeningitis,IgG4-RHP)是IgG4-RD累及颅内的一种表现形式,目前关于其发病率的研究鲜见,多为IgG4相关脑膜炎的个案报道[8,9]。IgG4相关脑膜病变临床表现不典型,从弥漫性症状(如慢性头痛、颈部僵硬、认知能力下降和癫痫发作)到神经压迫引起的局灶性机械症状(包括视神经病变、脑神经麻痹和皮质脊髓感觉运动缺陷)都可发生[10]。由于缺乏特征性影像学改变,故血清IgG4水平升高是诊断的重要线索,但IgG4-RHP确诊需要综合临床、影像学、血清学和病理组织学证据。IgG4-RHP的诊断目前主要依据IgG4-RD诊断指南[11]:① 1个或多个器官出现弥漫性或局限性肿胀,或肿块的临床表现;②血清IgG4浓度≥1 350 mg/L;③组织病理学检查,显著淋巴细胞、浆细胞浸润,席纹状纤维化及闭塞性静脉炎;IgG4+浆细胞浸润,IgG4+浆细胞/IgG+浆细胞>40%,且IgG4+浆细胞>10个/高倍视野。同时符合上述3项为确诊;同时符合①和③为很可能诊断;同时符合①和②为可能诊断。本例患者以颅脑病变为首发表现,并累及肺部、皮肤及骨骼,血清IgG4水平升高,且脑组织病理可见较多淋巴细胞和大量IgG4+浆细胞浸润,IgG4+浆细胞>60个/高倍视野,故IgG4-RD诊断明确。
IgG4-RD患者对糖皮质激素治疗反应良好。迄今为止,糖皮质激素仍是治疗IgG4-RD的一线药物,可用于疾病的诱导缓解和维持阶段[2,12]。IgG4-RHP的主要治疗方法与采用糖皮质激素治疗的IgG4-RD相似,通常口服泼尼松剂量为0.6~1.0 mg/(kg·d),持续2~4周,2~4周后逐渐减少并维持2.5~5.5 mg/d。对于糖皮质激素难治性患者,可以考虑使用免疫抑制剂,如硫唑嘌呤、甲氨蝶呤和环孢霉素。对于一些联合使用糖皮质激素和甲氨蝶呤无效或复发的病例,可改用利妥昔单抗治疗[13]。本例患者经甲泼尼龙冲击治疗、丙种球蛋白治疗和降颅压等对症支持治疗,症状较前好转。
作为一种跨多学科、累及全身多器官和系统的疾病,IgG4-RD临床表现多样、复杂,临床医师应高度关注该疾病,临床上遇到患者血清IgG4水平升高、病程较长、累及多系统,并排除肿瘤或感染等情况时,应高度怀疑此病,避免漏诊、误诊。
所有作者均声明不存在利益冲突

























