朗格汉斯细胞组织细胞增生症(LCH)临床上少见,合并肝功能损伤则更少见。本例患者长期间断发热、咳嗽、下肢疼痛,曾因骨痛行右股骨刮骨手术。后因食欲不振、尿黄就诊,发现患者有肝内胆汁淤积、肝脾大、肺多发囊性病变、骨质破坏、尿崩症,经多学科协作会诊,复诊股骨组织病理,最终诊断为多系统LCH。LCH起病隐袭、临床表现差异大,易被误诊或漏诊。现通过分享多学科协作诊断伴肝内胆汁淤积的多系统LCH的经过,为提高临床医师对LCH的认识和诊治水平提供参考。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
朗格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis, LCH)是一种以骨髓来源的树突状细胞中的朗格汉斯细胞异常增殖和浸润为主要特征的疾病,年发病率为(1~4.6)/100万,通常发生在儿童和青少年,高峰发病年龄为1~4岁,成人少见[1]。LCH可累及全身任何器官和部位,>2/3的病例仅累及骨骼和皮肤,合并肝功能损伤较少见[2]。LCH起病隐袭、临床表现差异大,易被误诊或漏诊。现通过1例伴肝内胆汁淤积的多系统LCH的多学科讨论,探讨LCH的诊治思路,以提高临床医师对LCH的认识和诊治水平。
患者男,45岁,因"间断咳嗽、发热15年,下肢疼痛12年,烦渴、多尿11年,食欲不振、尿黄1个月"于2017年2月16日被收治于河北医科大学第二医院。患者15年前无诱因出现咳嗽、发热,行胸部CT检查示弥漫性肺泡炎,经静脉输注青霉素(剂量不详)治疗1周后好转,此后间断咳嗽、发热,未予特殊诊治。12年前出现右股骨痛,髋部X线片示右股骨近段占位性病变,行穿刺活组织检查(以下简称活检)病理结果示嗜酸细胞肉芽肿。11年前出现烦渴、多饮、多尿,24 h尿量达10~20 L,因再次骨痛口服NSAID(布洛芬或萘普生,剂量和疗程不详),发现服药期间口渴减轻、尿量明显减少。2个月前再次出现咳嗽、咳痰,咳少量白色黏痰,伴低热,体温波动于37.5~38.0 ℃,多于夜间发热,无畏冷、寒战,无咽痛、盗汗,无胸闷、气短,无肌肉和关节疼痛,给予治疗(具体不详)后无缓解。1个月前患者无明显诱因出现食欲不振、恶心,厌食油腻、全身乏力,尿黄,皮肤、巩膜黄染,无腹痛、肝区不适,肝功能检查示TBil、ALP、GGT水平升高,腹部超声检查示肝脾大,口服利福平、多西环素、甘草酸二铵肠溶胶囊(剂量不详)治疗1个月后,仍有咳嗽、发热症状,进食量逐渐减至平时的1/3,且尿黄、皮肤和巩膜黄染加重,为求进一步诊治收入河北医科大学第二医院消化内科。患者30年前患左侧中耳炎,近2年反复复发。1年前患高血压,血压最高为160/100 mmHg(1 mmHg=0.133 kPa),未进行药物治疗和血压监测。患者未到过疫区和牧区,无牛羊和畜制品接触史,无吸烟或饮酒史,无类似家族史,肝损伤前无相关用药史。
体格检查示体温为36.8 ℃,脉搏为92次/min,呼吸频率为20次/min,血压为131/98 mmHg,BMI为20.76 kg/m2;神志清楚,皮肤、巩膜黄染,无肝掌、蜘蛛痣;头顶部可见散在红色丘疹;左外耳道可见脓性分泌物,左侧颈后胸锁乳突肌后缘可触及1枚大小约0.5 cm×0.5 cm的淋巴结,质硬,表面光滑,有压痛,活动度可;双肺呼吸音清晰,未闻及干湿性啰音;心率为92次/min,律齐,各瓣膜听诊区未闻及杂音;腹平坦,无压痛、反跳痛和肌紧张,肝脾未触及;右股部可见一长约10 cm的手术瘢痕。入院血常规示白细胞计数为9.59×109/L,中性粒细胞占比为0.799,血红蛋白为125 g/L,血小板计数为456×109/L。尿常规检查示尿比重值为1.004,尿渗透压为68 mOsm/(kg·H2O)。粪便常规、凝血常规检查均未见异常。ESR为70 mm/1 h,超敏CRP为104.8 mg/L。感染相关指标如结核分枝杆菌、布鲁氏菌、伤寒沙门菌等细菌或真菌均呈阴性。肝功能检查示TBil为82.00 μmol/L,DBil为44.4 μmol/L,白蛋白为31.2 g/L,ALT为25 U/L,AST为30.0 U/L,ALP为910 U/L,GGT为287 U/L,总胆汁酸为18.4 μmol/L;肾功能、电解质、心肌酶、血糖、血脂均未见异常。病毒性肝炎标志物均呈阴性。自身抗体[包括抗核抗体、抗干燥综合征A(Sjögren syndrome A,SSA)抗体等]、抗线粒体抗体(anti-mitochondrial antibody,AMA)、AMA-M2、风湿因子均呈阴性。免疫球蛋白(immunoglobulin,Ig)G、IgM、IgA均无异常,补体C3为2.02 g/L,补体C4为0.41 g/L。CA125为37.2 U/mL,铁蛋白为244.72 μg/L。甲状腺功能3项(促甲状腺激素、游离甲状腺素和游离三碘甲状腺原氨酸)检查均无异常。垂体激素6项(促甲状腺激素、促卵泡生成素、促黄体生成素、促肾上腺皮质激素、生长激素、泌乳素)检查示,促甲状腺激素为7.020 mU/L,泌乳素为19.76 μg/L,余4项指标均无异常。腹部超声提示肝大,肝回声增粗;肝右叶囊肿;脾大。胸部CT平扫检查示两肺内多发大小不等的囊性病变,部分融合,两肺内散在慢性炎症性病变;左侧第6肋骨局部小类圆形异常密度影(图1A),性质待定。肺功能检测示中度阻塞性通气功能障碍,轻度限制性通气功能障碍,重度弥散功能下降。上腹CT平扫和增强CT检查示肝实质内多发低密度影(图1B),性质待定,暂考虑囊肿可能性大;肝门区和腹膜后多个淋巴结,部分稍大;胆囊壁稍增厚,胆汁淤积。骶髂关节CT平扫检查示符合右股骨术后改变,右股骨骨质密度欠均匀;左侧髋臼和左股骨多发骨质破坏伴周围软组织影(图1C)。
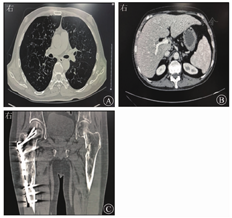
①LCH;②右股骨嗜酸细胞肉芽肿术后;③肺炎;④肝囊肿;⑤高血压2级(高危)。
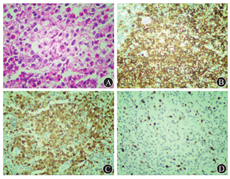
入院后嘱患者卧床休息,低盐、低脂饮食,口服熊去氧胆酸胶囊(0.25 g/次,3次/d)、静脉推注丁二磺酸腺苷蛋氨酸(1 g/次,1次/d)行保肝利胆治疗4周,静脉滴注哌拉西林他唑巴坦(4.5 g/次,2次/d)行抗感染治疗,静脉滴注氨溴索(30 mg/次,2次/d)行祛痰治疗2周,患者食欲不振、黄疸、咳嗽、咳痰等症状均减轻,体温维持在37 ℃左右。于2017年2月24日经血液内科、免疫风湿科、影像科、呼吸内科、骨科、内分泌科、皮肤科等多学科会诊,建议通过胸腔镜检查或开胸活检、骨骼病变穿刺或手术、肝脏穿刺、皮肤活检,或向河北医科大学第三医院(患者12年前曾于该院就诊)借阅病理切片行免疫组织化学等检查明确诊断。患者及其家属拒绝有创操作,选择向河北医科大学第三医院借阅病理切片,并于2017年3月16日复做H-E染色和免疫组织化学染色,结果示常规病理形态和免疫表型符合LCH,CD1α阳性(+),CD68阳性(+),溶菌酶散在阳性(+),S-100阳性(+),波形蛋白阳性(+),胎盘碱性磷酸酶(placental alkaline phosphatase,PLAP)阴性,Ki67阳性(为10%)(图2)。患者LCH诊断明确后,转血液内科进一步诊治,但因拒绝基因检测和化学治疗,于2017年3月22日自动出院。随访患者未行进一步检查和诊治,于2018年死亡。
消化内科张晓岚主任医师:根据患者病史总结病例特点如下:①患者为中年男性,30岁起病,病程长,多器官受损(包括弥漫性肺囊性病变、骨质破坏、尿崩症、肝脾和淋巴结大、头顶部皮疹、反复复发的中耳炎);②炎症指标ESR、超敏CRP明显升高;③感染、免疫相关指标均呈阴性;④CA125、铁蛋白轻度增高。其中,弥漫性肺囊性病变以间断咳嗽、咳痰、发热为首发表现,肺CT检查示两肺内多发、大小不等的囊性病变,部分融合,肺功能检测示混合性通气功能障碍,重度弥散功能下降;肝脾大、淋巴结大表现为食欲不振、厌食油腻,皮肤、巩膜黄染,肝功能检查提示胆汁淤积性肝损伤,影像学提示肝内多发低密度影,肝门区和腹膜后多个淋巴结部分稍大。可能病因分析如下。①成人斯蒂尔病:可表现为发热、肝脾大、淋巴结肿大、肝功能损伤,但一般不伴有肺囊性病变、骨质破坏和尿崩症。本例患者的抗核抗体、风湿因子均呈阴性,但无咽痛,白细胞计数不高,且伴有肺囊性病变、骨质破坏和尿崩症表现,故考虑成人斯蒂尔病可能性不大。②原发性胆汁性胆管炎合并干燥综合征:原发性胆汁性胆管炎易合并干燥综合征,可表现为发热、肝脾大、淋巴结肿大、肝功能损伤、肺囊性病变、皮疹。本例患者有肝内胆汁淤积表现,但抗核抗体、AMA、AMA-M2、抗SSA抗体、风湿因子均呈阴性,且无眼干、口干、反复腮腺肿大等干燥综合征表现,原发性胆汁性胆管炎合并干燥综合征可能性小。③脂质肉芽肿病:一种罕见的非LCH,多见于成年男性,可表现为骨痛、发热、肝脾大、肝功能损伤、尿崩症、肺间质病变等,需根据临床表现、影像学和病理学检查确诊,病理表现为含有泡沫样的荷脂细胞纤维炎症浸润,常伴有图顿巨细胞,CD1α、CD207阴性。④LCH:指CD1α阳性(+)和(或)CD207阳性(+)的树突状细胞异常克隆增生的一类疾病,可表现为肺囊性病变、骨质破坏、肝脾大、肝功能损伤、尿崩症、皮疹、耳部感染,最终需依靠病理学检查进行诊断,通常发生在儿童和青少年,成人少见。本例患者目前诊断尚不明确,并存在以下疑点。①患者肺脏、骨骼、肝脾影像学表现是否具有特异性,是否有助于疾病鉴别。②患者肝内胆汁淤积的原因是什么?是否与原发病相关?③患者存在尿崩症,其尿崩症是否与原发病有关?若有关,其发生机制是什么?如何解释患者口服NSAID后口渴减轻、尿量明显减少?④患者病史漫长,病情复杂,带给我们哪些反思和教训?因此,进行多学科讨论协助诊治。
影像科颜立群副主任医师:本例患者的影像学表现特点如下:①肺CT表现为两肺弥漫性囊性病变,大小不等、部分融合,主要在肺外围,不累及胸膜;②骨组织为多灶性溶骨性破坏,伴周围软组织影,似有葱皮样骨膜反应,无硬化表现;③肝脾、淋巴结CT表现为肝内多发低密度病变,脾脏未见异常,肝门区和腹膜后淋巴结大。根据患者病史和上述影像学表现,同意张晓岚医师关于LCH和脂质肉芽肿病诊断可能。患者两肺弥漫性囊性病变,主要位于肺外围,不累及胸膜,骨质破坏为溶骨性破坏,肝内多发低密度病变,淋巴结大,符合LCH影像学表现,不支持点为肝脏多发低密度病变强化期未见强化。脂质肉芽肿病也可出现肺囊性病变、溶骨性破坏、肝脏多发低密度病变、淋巴结大,但对称、平滑的小叶间隔增厚和局灶性胸膜下实变是其更常见的肺部CT表现;其骨骼受累的经典影像学表现为骨质硬化,偶有溶骨和硬化病变混合存在。综上考虑LCH可能性大。
免疫风湿科张明峰副主任医师:患者肺部CT提示弥漫性肺囊性病变。弥漫性肺囊性病变可见于感染性疾病、结缔组织病、肿瘤性疾病、遗传性疾病等。在免疫风湿科疾病中,肺部呈囊泡样改变的疾病以干燥综合征多见,但干燥综合征患者肺部呈萎缩样改变是由球蛋白升高所致的组织破坏,且病变很少累及骨骼,故本例患者暂不考虑干燥综合征诊断;IgG4相关性疾病亦可累及肺部,但也很少侵犯骨骼,故本例患者亦不考虑IgG4相关性疾病诊断;本例患者自身抗体阴性,IgG、IgM、IgA均无异常,故不考虑免疫风湿科疾病可能。
呼吸内科郭丽萍主任医师:患者为45岁男性,无吸烟史和遗传病家族史,临床表现为咳嗽、咳痰、间断发热,气短不明显,感染、免疫相关指标均呈阴性,肺功能检测示以阻塞性为主的通气功能障碍,伴重度弥散功能下降,肺部CT提示弥漫性薄壁囊腔,无毛刺样占位性病变、纵隔淋巴结、肺门淋巴结肿大等异常影像,可除外感染性疾病、结节病、肺腺癌肺内转移、淋巴管肌瘤、淀粉样变性、窦组织细胞增生症伴巨大淋巴结病等疾病;结合患者存在肝功能损伤、骨质破坏、尿崩症,既往股骨组织病理提示嗜酸细胞肉芽肿,考虑LCH可能性大。15%的LCH患者会出现肺部受累,且肺部受累多见于年轻成人吸烟者,而儿童罕见。日本的研究数据表明,女性肺LCH的患病率为0.07/10万,男性为0.27/10万[3]。临床表现为慢性咳嗽,以干咳为主,活动后气短和呼吸困难,可伴胸痛、发热、咯血等,甚至可发生气胸,随着病情进展出现肺动脉高压和右心力衰竭。约1/5的患者不伴肺功能障碍,其余病例表现为以阻塞性为主的通气功能障碍,伴弥散功能障碍,以一氧化碳弥散能力减弱最为常见。6 min步行测试有助于观察患者活动时的缺氧情况。肺LCH患者不同时期胸部影像学改变不一:早期,肺部受累的CT表现为两肺内广泛分布的细支气管周围渗出的小斑片影、磨玻璃影和小结节影,部分可融合成片状,以肺外围为主;中晚期,CT表现为肺内形态各异、大小不等的纤维条索影和壁薄而光整的圆形或类圆形囊状影,囊腔融合出现蜂窝肺,病变一般不累及胸膜。正电子发射计算机断层显像-计算机断层扫描有助于早期发现肺部受累的结节样改变[4]。依靠临床影像分析诊断肺LCH的诊断率可达84%。支气管肺泡灌洗液中表达CD1a抗原的细胞升高5%以上,结合影像学改变更有助于肺LCH的诊断[4]。鉴于肺LCH经皮肺穿刺和经支气管镜肺活检的检出率低,建议行胸腔镜检查或开胸活检以明确诊断。
骨科曹舒兴主治医师:患者目前发现的骨骼受累部位为股骨、肋骨,为溶骨性破坏,伴有周围软组织肿胀,既往右股骨手术病理提示嗜酸细胞肉芽肿。80%的LCH患者出现骨骼浸润,骨骼受累特点为溶骨性破坏,主要部位为中轴和近端骨。而脂质肉芽肿病骨骼受累主要特点为骨质硬化,几乎所有脂质肉芽肿病患者都伴有对称性的下肢长骨骨硬化症。故本例患者骨质破坏考虑由LCH所致。患者术后再次出现骨痛,CT检查发现新发左侧股骨溶骨性病变,提示疾病复发,患者存在其他器官受累,可嘱患者减少活动,待明确诊断后行化学治疗。
内分泌科王绵主任医师:患者目前存在烦渴、多饮、多尿,尿比重、渗透压减低,符合尿崩症临床表现。患者每日尿量可达10~20 L,尿比重、渗透压明显减低,且患者无肾脏疾病病史,肾功能检查无异常,考虑中枢性尿崩症可能性大,可行禁水试验以明确为中枢性或肾性尿崩症,但患者表示不可耐受,因此,可尝试弥凝诊断性试验(去氨加压素一般对肾性尿崩症无效)和垂体MRI检查(垂体后叶T1高信号消失提示中枢性尿崩症)鉴别尿崩症类型。LCH和脂质肉芽肿病均可出现中枢性尿崩症(国外LCH合并中枢性尿崩症发生率为29.6%,国内为5%;脂质肉芽肿病合并中枢性尿崩症发生率为28%),其发病机制为组织细胞在下丘脑、垂体柄、垂体后叶区异常增殖和浸润,导致抗利尿激素合成、转运和分泌不足。LCH垂体MRI表现为垂体柄增粗,脂质肉芽肿病MRI表现为下丘脑、垂体柄、垂体后叶区肿大或异常强化。综合患者病史,考虑患者的尿崩症与原发病相关。由于垂体活检操作难度和风险较高,建议行肺或骨组织活检以明确诊断。患者目前口服NSAID后口渴减轻、尿量明显减少,可能机制如下:①影响下丘脑的功能,具体机制尚不清楚;②抑制肾脏水通道蛋白从细胞膜表面脱离,从而抑制对水的转运;③抑制前列腺素的合成,从而收缩肾脏血管,减少尿量。
皮肤科佟盼琢主任医师:本例患者头顶皮疹反复存在、迁延不愈,间断有黄色渗液,目前皮疹表现为散在的头皮红色丘疹,无渗液,符合脂溢性皮炎表现,但结合患者多器官受损,不能除外引起多系统受累的疾病所致皮损。LCH可表现为头皮红色丘疹,也可表现为脂溢性皮疹或溃疡性病变。脂质肉芽肿病最常见的皮肤表现为黄斑瘤,好发于眼睑,虽可表现为丘疹,但是颜色多为黄色或红棕色并融合成斑块。可留取头皮皮疹病理以证实。
消化内科白文元主任医师:患者为肝内胆汁淤积性肝损伤,常见病因筛查均呈阴性,包括无饮酒史,无肥胖、遗传病病史,肝损伤前无相关用药史,血糖、血脂均无异常,病毒性肝炎标志物和自身抗体均呈阴性,可排除酒精性或非酒精性肝病、病毒性肝炎、自身免疫性肝病、结缔组织病致胆汁淤积、药物性肝损伤;结合患者存在多器官受损,ESR明显增快,CA125、铁蛋白增高,考虑肿瘤性疾病所致肝损伤可能性大。有研究显示约15%的LCH患者出现肝脏受累,其临床表现无特异性,肝功能指标TBil、ALP、GGT、ALT、AST均可升高,且LCH所致肝功能损伤表现为胆汁淤积者占64%~74%。LCH浸润肝脏时倾向于浸润胆管[2],10%~15%的LCH患者出现硬化性胆管炎。本例患者肝功能指标TBil、ALP、GGT明显升高,CT平扫检查示肝脏多发低密度病变,以上结果符合LCH肝脏受累表现;但本例患者CT强化期低密度病变未显示强化,未见门静脉周围晕征和肝纤维化等LCH肝脏影像学表现[5],尚不能明确诊断,需与脂质肉芽肿病相鉴别。脂质肉芽肿病致肝脏受累罕见,肝功能生物化学检查亦无特异性。有零星报道显示,脂质肉芽肿病肝脏受累的CT表现为多发低密度结节状病变(最大径为0.5~3.2 cm)或肝纤维化表现。综合多学科意见,考虑本例患者为LCH可能性大,建议行肝脏穿刺活检以明确诊断。若患者及其家属不同意进行胸腔镜检查或开胸活检、骨骼病变穿刺或手术、肝脏穿刺、取皮疹病理等措施,可借阅病理切片,并切白片进一步行免疫组织化学(如CD1α、CD68、Ki67、CD45、溶菌酶、S-100相关指标)以明确诊断。治疗方面,由于患者食欲不振、黄疸较前减轻,肝功能无加重情况,咳嗽、咳痰减轻,体温在37 ℃左右,可继续保肝、抗感染、化痰治疗,待明确诊断后转血液内科行进一步治疗。
血液内科郭晓楠主任医师:患者及其家属拒绝有创操作,故向河北医科大学第三医院借阅病理切片并复做H-E染色和免疫组织化学染色。H-E染色示大量朗格汉斯细胞浸润,伴散在嗜酸性粒细胞;免疫组织化学染色示,CD1α阳性(+),CD68阳性(+),溶菌酶散在阳性(+),S-100阳性(+),波形蛋白阳性(+),PLAP阴性,Ki67阳性(为10%)。根据2013年国际组织细胞协会修订的LCH诊断标准,在光学显微镜下见典型的朗格汉斯细胞的基础上,CD1α阳性和(或)CD207阳性,可确诊LCH。LCH临床表现差异大,可表现为肺部感染、溶骨性破坏、肝功能损伤、脾大和(或)脾功能亢进、尿崩症、脑部症状、肠道病变、血象三系(红细胞、白细胞和血小板)减少、皮疹、耳部感染等[6]。该患者最早出现的症状是中耳炎,其发生可能与LCH相关,有研究显示4%~61%的LCH累及耳颞部,最初的临床表现可能仅为耳渗液、流脓或耳颞肿物,此后才出现耳渗血、耳痛、耳鸣、头晕、听力下降等。按病损范围LCH可分为单系统LCH和多系统LCH,累及肝、脾或造血系统者为高危LCH。本例患者骨髓穿刺检查示刺激性骨髓象,未见受累;患者肝脾肿大,尽管患者拒绝肝脏穿刺活检,导致肝脏或脾脏穿刺检查的病理依据缺乏,但患者存在多器官受损,其中肝损伤为肝内胆汁淤积,CT平扫检查示肝脏多发低密度病变,且未找到肝脾肿大的其他病因,因此考虑LCH累及肝脾可能性大,故本例患者应诊断为多系统、高危LCH。
LCH的发病机制目前倾向于肿瘤增生性假说,主要与MAPK通路异常活化相关。50%以上LCH存在丝氨酸/苏氨酸激酶v-RAF鼠肉瘤病毒癌基因同源物B1(the serine threonine kinase v-RAF murine sarcoma viral oncogene homologue B1,BRAF)位点V600E突变,另外还发现丝裂原活化蛋白激酶激酶1(mitogen-activated protein kinase kinase 1,MAP2K1)、丝氨酸/苏氨酸激酶v-RAF鼠肉瘤病毒癌基因同源物A(the serine threonine kinase v-RAF murine sarcoma viral oncogene homologue A,ARAF)、禽红细胞白血病病毒癌基因同源物3(avian erythroblastic leukemia viral oncogene homolog 3,ERBB3)、神经母细胞瘤RAS病毒癌基因同源物(neuroblastoma RAS viral oncogene homolog,NRAS)等基因突变[7,8]。进行基因检测有助于制定治疗方案。目前LCH治疗方法包括手术、联合化学治疗、靶向治疗、造血干细胞移植等。LCH治疗选择必须根据疾病范围和严重程度进行调整,并考虑死亡风险。对于成人肺LCH的治疗,首要任务是戒烟;对于不吸烟者或戒烟后病情进展的患者,可以选择糖皮质激素或克拉屈滨的全身治疗;对于极具侵袭性的肺LCH合并不可逆肺损伤或严重肺动脉高压的患者,可以考虑肺移植;对于仅皮肤或骨骼受累的单系统LCH患者,推荐局部治疗,包括手术、局部激素、口服甲氨蝶呤或沙利度胺;对于多系统LCH或合并多灶性骨病变的单系统LCH患者,强烈推荐全身治疗;对于LCH合并多系统疾病的患者,长春新碱联合甲泼尼龙方案是一线治疗方案;对于复发难治性LCH患者;可采取分子靶向治疗(维罗非尼、达拉非尼治疗)、造血干细胞移植[9,10,11]。本例患者为多系统LCH,无吸烟史,病情不断进展,推荐长春新碱联合甲泼尼龙方案,但患者拒绝化学治疗,自动出院。随访患者未进一步诊治,最后死亡。
本例患者12年前出现右股骨痛,经病理检查证实为嗜酸细胞肉芽肿,但未予重视,也忽视了30年前的中耳炎病史。此后,患者反复出现咳嗽、发热,亦未考虑与LCH相关。患者因肝功能损伤就诊于河北医科大学第二医院,多学科协作会诊使得患者诊断得以明确。成人LCH发病隐袭,通常为慢性病程,早期可能仅发现单系统受损,且LCH临床表现差异较大,极易被误诊、漏诊。LCH伴肝功能损伤的情况虽较少见,但是对于肝功能异常、肝脾肿大合并肺囊性病变、骨质破坏、中枢性尿崩症等多器官受损患者,要考虑LCH的可能性,注意以一元论解释疾病。目前对于LCH的治疗特别是成人LCH的治疗仍具有挑战性,需要充分认识LCH,及早识别疾病,以改善患者的临床预后。
所有作者均声明不存在利益冲突


























