
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
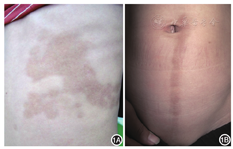
患儿男,9岁,左胸腹部不规则褐色硬化斑4年,脐中线带状萎缩斑2年。皮损无明显自觉症状,近期无明显扩大,起病前无外伤、注射、炎症及服药史。患儿既往体健,家族中无类似疾病史。体检:一般情况可,各系统检查未见明显异常。皮肤科检查:左胸腹部见片状融合性褐色斑片,形态稍不规则,呈"S"形走向(图1A),大致沿Blaschko线分布,触之较硬;脐中线处见纵性带状分布萎缩斑,伴轻微色素沉着,无明显硬化,大致沿Blaschko线分布(图1B)。脐中线处皮损活检:基底层色素增加,轻度乳头瘤样增生,真皮胶原纤维大致正常,真皮浅层血管周围少许淋巴细胞浸润(图2A);左侧腹部皮肤活检:角化过度,真皮内局部胶原增生和玻璃样变,毛囊数减少,真皮浅中层血管及附属器周围淋巴细胞浸润(图2B)。弹性纤维染色均未见明显异常。实验室检查:血、尿、便常规及肝肾功能未见异常。诊断:Moulin线状皮肤萎缩伴局限性硬皮病。治疗:口服脉管复康片1.2 g每日3次、复方丹参片0.64 g每日3次,多磺酸黏多糖乳膏和维生素E乳膏外用,治疗3个月后皮损外观及质地改善不明显,目前仍在随访中。
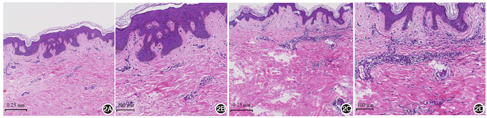
Moulin线状皮肤萎缩(linear atrophoderma of Moulin)病因尚不确切,好发于儿童及青少年,典型临床特点为沿Blaschko线分布的萎缩性色素沉着斑,常单侧分布,一般不累及其他系统。组织病理示表皮基底色素增加,棘层不规则增厚,真皮胶原纤维和弹性纤维基本正常,真皮浅层血管周围少量淋巴细胞浸润[1]。局限性硬皮病,是一种特发性炎症性疾病,其中线状硬斑病是儿童硬斑病中最常见的亚型[2],表现为线状分布的硬斑型斑块,其色素变化较其他类型硬皮病更明显。本例患儿两处皮损形态相似,均为带状分布的皮肤萎缩斑伴色素沉着,其中脐中线部位的皮损组织学主要表现为表皮基底色素增加,真皮胶原纤维和弹性纤维基本正常,真皮萎缩不明显,根据上述特点,该部位皮损诊断为"Moulin线状皮肤萎缩"。皮下组织变薄可能是导致其临床表现为萎缩的原因,有学者通过超声检查证实了上述假说[3]。而左侧胸腹部皮损的病程相对稍长,且出现硬化,组织学检查同时可见真皮内局部胶原增生、玻璃样变,毛囊数减少,真皮浅中层血管及附属器周围淋巴细胞浸润,更符合"局限性硬皮病"的诊断。结合本例患儿皮损的特殊性及疾病发展过程,我们推测Moulin线状皮肤萎缩可能是硬皮病的特殊类型或者顿挫型,故尚不除外脐中线部位皮损发展为硬皮病的可能,需密切随访。
总体来讲,Moulin线状皮肤萎缩和硬皮病在临床及病理表现方面是类似且相互重叠的,因此有学者认为上述疾病属于同一疾病谱系[4],甚至有学者认为"Moulin线状皮肤萎缩"即沿Blaschko线分布的"线状硬皮病"。然而目前关于二者发病机制的研究甚少,其中可能涉及多种因素,包括自身免疫、遗传,以及血管功能障碍等等。国外学者认为疾病沿Blaschko线分布可能系胚胎发生早期的体细胞突变所致,这种皮损分布可能与遗传学的镶嵌现象相关[5]。本例患儿同时具有Moulin线状皮肤萎缩及局限性硬皮病的表现,且两者均沿Blaschko线分布,支持上述观点。此外,二者的组织病理学亦有相似之处,即血管周围淋巴细胞为主的炎症浸润,推测其发病机制和免疫异常相关。既往曾有文献报告1例28岁男性Moulin线状皮肤萎缩患者,其血清免疫指标异常,且组织学可见黏蛋白沉积等结缔组织疾病的变化,推测其发病可能与免疫异常有关,或者本身即是一种结缔组织病[1]。而有些硬斑病患者,特别是泛发性或线状变异型患者中,自身抗体水平增加,且其伴发自身免疫性疾病的概率升高[6]。综上,我们推测二者为同一起源,其具体发生机制以及两者之间的关联性值得进一步研究。
所有作者均声明不存在利益冲突

























